Capítulo 5
Metabolómica para la ciencia básica
En este capítulo, aprenderás cómo la metabolómica puede esclarecer mecanismos que pasan desapercibidos para otras ciencias «ómicas» con el fin de transformar las estrategias terapéuticas futuras; cómo ha revelado conocimientos clave que han mejorado significativamente nuestra comprensión y las implicaciones clínicas del eje microbioma-cerebro y la salud de la población; y cómo puede generar datos significativos a partir de diversas matrices de muestras que no suelen analizarse habitualmente.
Visión general
La metabolómica ha sido fundamental para avanzar en nuestra comprensión de los principios de la ciencia básica, gracias a la profunda perspectiva que ofrece sobre los mecanismos que influyen directamente en el fenotipo y la historia natural de las enfermedades, así como a su capacidad para impulsar descubrimientos y hallazgos que generan hipótesis. En los últimos años, también ha contribuido de manera significativa a temas de especial interés, como la investigación centrada en el microbioma y la salud de la población. Por último, su compatibilidad con matrices de muestras poco comunes la hace ampliamente aplicable a la mayoría de las áreas de las ciencias de la vida. A continuación, destacamos algunos estudios que muestran cómo la metabolómica puede esclarecer mecanismos y aplicarse a tipos de muestras alternativos.
Mecanismo
Remodelación cardíaca en la insuficiencia cardíaca
Introducción. La insuficiencia cardíaca (IC) se asocia a una disminución de la flexibilidad metabólica, por lo que el corazón no puede aumentar el metabolismo de la glucosa en respuesta al estrés, lo que da lugar a una reducción de la eficiencia cardíaca y a la progresión de la IC. En algunas circunstancias, la función sistólica del ventrículo izquierdo (VI) puede recuperarse si existe miocardio viable y se trata con éxito la causa subyacente de la IC. La terapia de resincronización cardíaca (TRC) es un tratamiento para la IC en el que un bloqueo de rama izquierda amplio provoca una contracción desincronizada entre el tabique y la pared lateral del VI. La TRS mejora de forma aguda la hemodinámica cardíaca y la eficiencia del oxígeno, y se observa una mejora adicional de la función del VI a medida que se produce la remodelación con el tiempo. Los mecanismos que relacionan la TRS con la remodelación cardíaca estructural a largo plazo siguen sin estar claros. Se ha demostrado que la TRS modifica el metaboloma circulante con el tiempo, pero no está claro si esto es causa o efecto.
Datos preliminares y objetivos del estudio. En última instancia, una mejora de la función contráctil requiere un mayor suministro de ATP al aparato contráctil. Por lo tanto, es plausible que el grado de flexibilidad metabólica que conserva el corazón con insuficiencia pueda ser clave para su capacidad de remodelación. El objetivo de este estudio fue poner a prueba esta hipótesis caracterizando el grado de remodelación ventricular izquierda en respuesta a 6 meses de TRC y determinar si la TRC puede alterar de forma aguda la captación de sustratos alejándola del fenotipo metabólico de la IC para, en última instancia, favorecer la remodelación [16].
Métodos. En este estudio se analizó a pacientes con insuficiencia cardíaca junto con grupos de comparación adecuados, utilizando fenotipado clínico y perfiles metabolómicos globales. Tras un ayuno nocturno, se estabilizó a los pacientes con una infusión de insulina y glucosa durante al menos una hora antes de la implantación del dispositivo de resincronización cardíaca. Se realizaron mediciones del bucle presión-volumen y se obtuvieron muestras arteriovenosas coronarias. Se interrumpió la infusión de insulina/glucosa y se inició una infusión de intralípidos durante 15 minutos, tras lo cual se repitieron las mediciones. Las muestras de sangre arterial y venosa se analizaron mediante metabolómica no dirigida, y la flexibilidad metabólica se evaluó mediante la valoración de la captación de determinados metabolitos y el cálculo de la eficiencia del oxígeno miocárdico. Se utilizaron análisis estadísticos y de vías metabólicas para identificar los metabolitos asociados a una flexibilidad metabólica alterada.
Resultados. La TRC provocó un aumento significativo de la captación de ácidos grasos no esterificados (AGNE), sin que se observara un aumento significativo de la captación de glucosa, beta-hidroxibutirato o lactato (Figura 29). Se observó una fuerte correlación entre la mejora aguda del rendimiento hemodinámico cardíaco inducida por el acortamiento del QRSd debido a la TRC. Se observó una fuerte correlación positiva entre el aumento del trabajo sistólico y la captación de AGNE. El cambio en la captación de sustratos en respuesta a la TRC se correlacionó con la remodelación inversa a largo plazo del volumen telediastólico del ventrículo izquierdo (Figura 30). Se observó una correlación significativa entre el aumento de la captación de AGNE y la reducción del VTDVI. El análisis lipidómico mostró que esto se debía a aumentos tanto en los ácidos grasos de cadena larga como en los de cadena media.
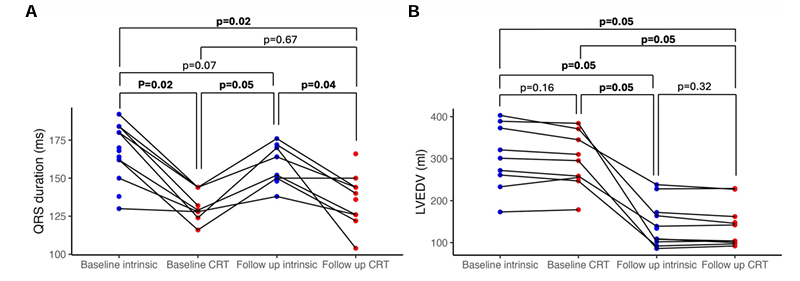
Figura 29. Remodelación crónica del ventrículo izquierdo tras 6 meses de TRC, independientemente de los cambios agudos en la duración del QRS. Duración del QRS (A, 13 pacientes) y volumen telediastólico del ventrículo izquierdo (B, 9 pacientes) en el momento del implante y en el seguimiento a los 6 meses, con ritmo intrínseco y estimulación optimizada mediante terapia de resincronización cardíaca. Imagen reproducida de Green et al., Eur Heart J, 2025, con licencia CC BY 4.0.
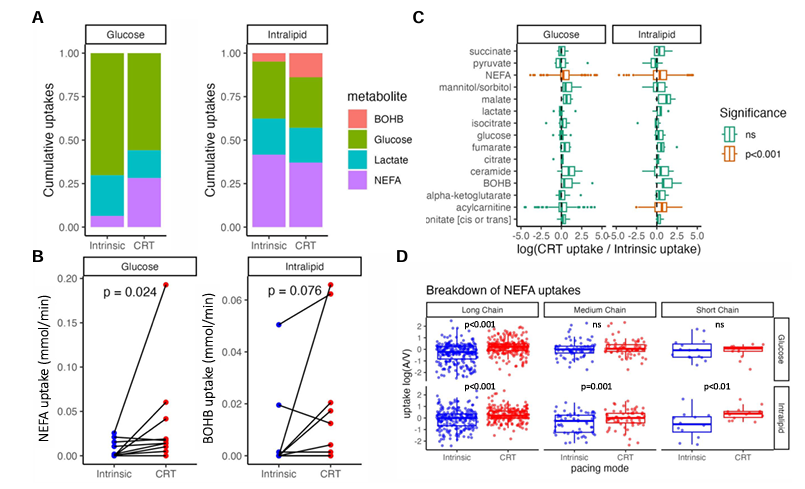
Figura 30. Efecto de la TRC sobre la captación de sustratos cardíacos durante infusiones de insulina/glucosa e intralípidos. (A) Proporción acumulada de captación de AGNE, beta-hidroxibutirato, lactato y glucosa durante la conducción intrínseca y la TRC optimizada, mientras se administran infusiones de intralípidos e insulina/glucosa. (B) Ilustración del efecto de la terapia de resincronización cardíaca sobre la captación de ácidos grasos no esterificados y β-hidroxibutirato durante infusiones de insulina/glucosa e intralípidos, respectivamente. (C) Análisis metabolómico que ilustra la diferencia en la captación de metabolitos intermedios inducida por el inicio de la terapia de resincronización cardíaca, donde los valores positivos indican un aumento de la captación con la terapia de resincronización cardíaca. (D) Análisis lipidómico que ilustra el efecto de la terapia de resincronización cardíaca sobre las captaciones [definidas como log (arterial/venosa)] de ácidos grasos no esterificados de diferente longitud de cadena durante infusiones de insulina/glucosa e intralípidos. Imagen reproducida de Li et al., J Exp Med, 2014, con licencia CC BY 4.0
Conclusiones del estudio
- Estos hallazgos demuestran que, en la miocardiopatía no isquémica, el corazón conserva una flexibilidad metabólica considerable y, por lo tanto, la terapia de reestructuración cardíaca (CRT) es capaz de revertir el fenotipo metabólico de la insuficiencia cardíaca hacia un fenotipo más fisiológico de captación de ácidos grasos libres.
- El grado de flexibilidad metabólica conservada está relacionado con la remodelación inversa a largo plazo del volumen telediastólico del ventrículo izquierdo.
- En este estudio, la metabolómica reveló que la remodelación cardíaca en respuesta a la TRC podría deberse a cambios en el metabolismo celular provocados por la TRC, más que a los efectos agudos del estrechamiento del QRSd.
Identificación de una nueva diana terapéutica para la nefropatía diabética
Introducción. La enfermedad renal diabética (ERD) es una de las principales causas de enfermedad renal crónica y de insuficiencia renal. Aunque los inhibidores del cotransportador de sodio-glucosa tipo 2 (SGLT2) se desarrollaron inicialmente para reducir la glucemia mediante el aumento de la excreción urinaria de glucosa, los ensayos clínicos han demostrado que también ofrecen una sólida protección renal y cardiovascular, incluso en pacientes sin diabetes. Sin embargo, los mecanismos biológicos responsables de estos efectos protectores siguen sin estar claros, sobre todo porque el SGLT2 se expresa únicamente en las células tubulares proximales (CTP) del riñón, lo que plantea la cuestión de cómo el hecho de actuar sobre un transportador en una población celular limitada puede mejorar la función renal general.
Datos preliminares y objetivos del estudio. Estudios previos sugieren que los riñones de los pacientes diabéticos presentan alteraciones metabólicas sustanciales, entre ellas un aumento de la glucólisis y una disfunción mitocondrial, que la inhibición de SGLT2 podría normalizar. Por lo tanto, los investigadores del estudio plantearon la hipótesis de que la pérdida o la inhibición de SGLT2 altera las vías metabólicas dentro de las células renales para producir metabolitos que protegen el riñón. El objetivo de este estudio fue comprobar esta hipótesis mediante análisis metabolómicos, transcriptómicos y epigenéticos para identificar los cambios metabólicos en las células tubulares primarias (PTC) que carecen de la función de SGLT2 y determinar si estas vías contribuyen a la protección renal [17].
Métodos. Se alimentó a ratones machos de tipo salvaje (WT) y ratones con pérdida de función de SGLT2 «Sweet Pee» (SP) con una dieta normal o una dieta rica en grasas (HFD) durante un máximo de 18 semanas para inducir estrés metabólico y enfermedad renal diabética en fase temprana. Se evaluaron la función renal, la histología y los parámetros metabólicos midiendo la creatinina sérica y la relación albúmina/creatinina en orina, y mediante inmunotinción del tejido renal para detectar marcadores de lesión y fibrosis. Se realizó secuenciación de ARN unicelular (scRNA-seq) en células tubulares proximales, y se llevó a cabo un perfil metabolómico no dirigido en la corteza renal y el suero. Se realizó un perfil epigenético CUT&RUN para medir los cambios en la metilación de histonas asociados a los niveles alterados de metabolitos.
Resultados. En ratones alimentados con una dieta rica en grasas, aquellos que carecían de la función de SGLT2 mostraron menos daño renal, inflamación, fibrosis y proteinuria que los ratones de tipo salvaje, a pesar de un estrés metabólico similar. La secuenciación de ARN unicelular identificó una población de células tubulares proximales dañadas que aparecía en los riñones de tipo salvaje en condiciones de dieta rica en grasas, pero que se suprimía en gran medida en ausencia de la función de SGLT2. El análisis metabolómico reveló que los riñones que carecían de SGLT2 presentaban una mayor actividad del metabolismo de la metionina, en particular niveles más elevados del metabolito S-adenosilmetionina (SAM) (Figura 31). Los experimentos funcionales demostraron que el bloqueo de la enzima MAT2A, productora de SAM, eliminaba los efectos protectores de la pérdida de SGLT2, mientras que la suplementación con SAM reducía las respuestas inflamatorias en células renales cultivadas expuestas a estrés metabólico (Figura 32). La inhibición farmacológica de SGLT2 produjo efectos protectores similares a los de la pérdida genética de SGLT2. Por último, los análisis epigenéticos mostraron que los niveles elevados de SAM aumentaban la metilación represiva de histonas (H3K27me3) en los promotores de genes inflamatorios, incluidos los de la vía NF-κB, lo que conducía a una reducción de la señalización inflamatoria. En conjunto, estos resultados indican que la inhibición de SGLT2 promueve la protección renal al aumentar la represión epigenética mediada por SAM de las vías inflamatorias durante el estrés metabólico.
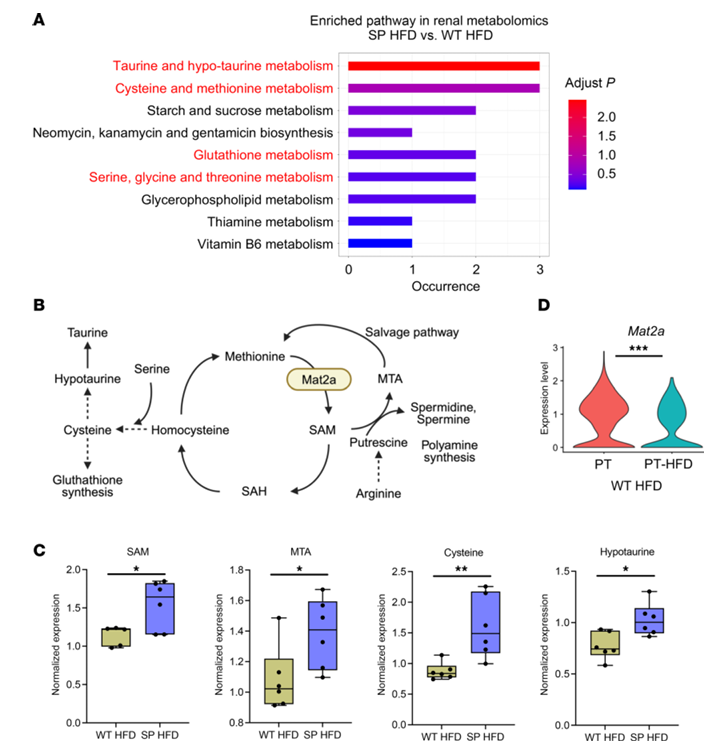
Figura 31. Perfiles metabólicos en el riñón. (A) Análisis de vías de enriquecimiento metabólico en tejido aislado de la corteza renal. El color indica el valor P ajustado. (B) Metabolismo de la metionina y vías de la red. (C) Expresión relativa de SAM, MTA y metabolitos relacionados con el metabolismo de la cisteína y la taurina en la corteza renal (n = 6 por grupo). Los rectángulos muestran los percentiles 25 a 75, las líneas centrales indican las medianas, las barras se extienden hasta el mínimo y el máximo, y se muestran todos los puntos de datos. (D) Gráfico de violín que muestra la expresión de Mat2a en el túbulo proximal en células normales del túbulo proximal (PT) y en la subpoblación de PTC (PT-HFD) que surgió en ratones de tipo salvaje alimentados con una dieta rica en grasas (WT HFD). Imagen reproducida de Maekawa et al., J Clin Invest, 2025, con licencia CC BY 4.0.
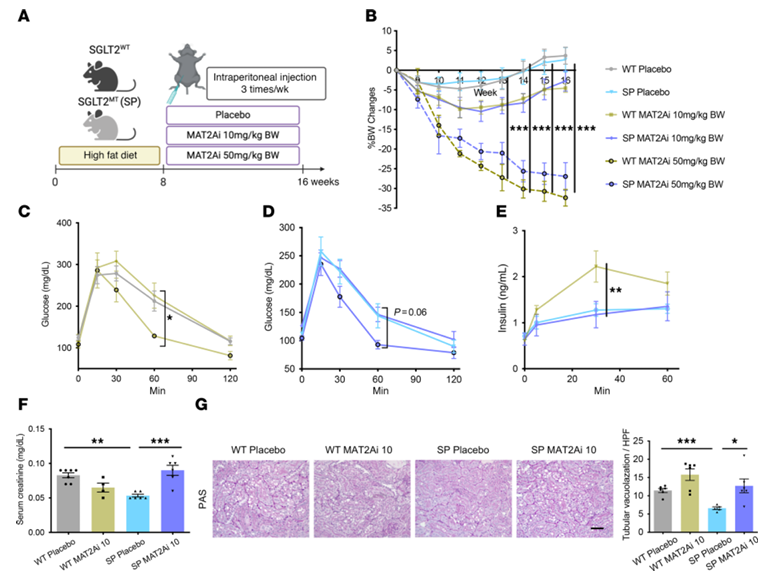
Figura 32. La inhibición de la enzima metionina, MAT2A, anula la protección renal en ratones con SPHFD. (A) Protocolo esquemático. (B) Evolución cronológica del porcentaje de variación del peso corporal. (C–E) Una dosis baja de MAT2Ai no altera la tolerancia a la glucosa ni la capacidad de secreción de insulina, pero una dosis alta de MAT2Ai sí las reduce. (F) Nivel de creatinina sérica. (G) Imágenes representativas de la tinción PAS. Imagen reproducida de Maekawa et al., J Clin Invest, 2025, con licencia CC BY 4.0
Conclusiones del estudio
- Este estudio aporta pruebas de que los metabolitos desempeñan un papel regulador activo en la progresión de la enfermedad renal crónica y sugiere que actuar sobre el metabolismo de la metionina o las vías relacionadas con el SAM podría constituir una estrategia terapéutica viable para los pacientes que no toleran los inhibidores de SGLT2.
- La metabolómica resultó fundamental para caracterizar la relación entre la inhibición de los SGLT2, el metabolismo renal y la respuesta inflamatoria en ratones, lo que puede servir de punto de partida para futuros estudios en humanos.
Identificación de un posible tratamiento preventivo para un tumor maligno agresivo
Introducción. El adenocarcinoma ductal pancreático (PDAC) es la tercera causa principal de muerte por cáncer en todo el mundo y uno de los pocos tipos de cáncer cuya prevalencia está aumentando a nivel global. En la actualidad, la tasa de supervivencia a cinco años se sitúa en el 13 %, debido en gran medida a que la mayoría de los diagnósticos se producen en una fase avanzada, cuando el tumor ya ha hecho metástasis y la resección ya no es una opción. Más del 90 % de los tumores de PDAC contienen mutaciones activadoras de KRAS, lo que convierte a KRAS en un factor clave del crecimiento tumoral y en una diana terapéutica atractiva. Sin embargo, las respuestas clínicas a los inhibidores de KRAS y a los fármacos dirigidos a la vía de señalización de KRAS (como los inhibidores de RAF/MEK/ERK) han sido limitadas, con respuestas solo parciales y un rápido desarrollo de resistencia a los fármacos. La mayoría de las estrategias de combinación actuales se centran en componentes de la vía de señalización canónica de KRAS, pero las células cancerosas pueden eludir estas dianas mediante mecanismos compensatorios, lo que pone de relieve la necesidad de identificar nuevas vulnerabilidades fuera de la vía tradicional de KRAS.
Datos preliminares y objetivos del estudio. Para dar respuesta a esta necesidad no cubierta, los investigadores del estudio se propusieron identificar los genes esenciales para la supervivencia de las células del PDAC con mutación en el gen KRAS, con el objetivo de descubrir nuevas dianas terapéuticas y estrategias combinadas que pudieran mejorar la eficacia de los inhibidores de KRAS en el cáncer de páncreas [18].
Métodos. Se utilizaron cribados de inactivación de genes mediante CRISPR/Cas9 a escala genómica, conjuntos de datos de interferencia de ARN y grandes bases de datos de dependencia del cáncer para identificar los genes que son esencialmente necesarios para las células de PDAC con mutación en KRAS. Se llevaron a cabo cribados de sensibilidad a fármacos para identificar compuestos que inhibieran preferentemente las células con mutación en KRAS. Los genes candidatos se validaron mediante técnicas de silenciación o eliminación génica y ensayos de viabilidad celular en líneas celulares de PDAC. Se utilizaron secuenciación de ARN, metabolómica, proteómica y trazado de isótopos estables para analizar los cambios en el metabolismo de los nucleótidos y las vías celulares tras la inhibición génica o el bloqueo de KRAS. La importancia funcional de las dianas se evaluó además utilizando modelos de ratones modificados genéticamente y modelos de trasplante ortotópico de tumores. Se utilizaron compuestos PROTAC (quimeras dirigidas a la proteólisis) que degradan selectivamente IMPDH2 para evaluar su potencial terapéutico en organoides derivados de pacientes y modelos de tumores en xenoinjertos.
Resultados. Se identificó la biosíntesis de novo de nucleótidos de guanina (DNGB) como una vulnerabilidad metabólica clave en las células de PDAC con mutación en KRAS, siendo el gen crítico el IMPDH2, que codifica una enzima esencial para la DNGB. La inhibición farmacológica de la actividad de IMPDH2 o de otras enzimas de la DNGB redujo significativamente el crecimiento de las células de PDAC. En modelos murinos, la deleción genética de Impdh2 redujo la iniciación de tumores pancreáticos, ralentizó la progresión de la enfermedad y mejoró la supervivencia (Figura 33). Los análisis metabolómicos mostraron que el KRAS mutante estimula la biosíntesis de purinas para aumentar preferentemente la producción de nucleótidos de adenina frente a los de guanina, creando un «cuello de botella» metabólico para la síntesis de guanina (Figura 34). Como resultado, las células del PDAC se vuelven dependientes de IMPDH2 para mantener los niveles de nucleótidos de guanina y seguir proliferando. La inhibición de IMPDH2 agotó los nucleótidos de guanina, lo que afectó al crecimiento de las células del PDAC. Es importante destacar que se demostró que la expresión de IMPDH2 no estaba regulada por la vía de señalización canónica de KRAS, lo que significa que las células cancerosas no podían compensar la inhibición de IMPDH2 a través de la señalización de KRAS. Un compuesto selectivo que degrada la IMPDH2 (MED-B-4) suprimió el crecimiento de las líneas celulares de PDAC con mayor potencia que los inhibidores de IMPDH existentes.
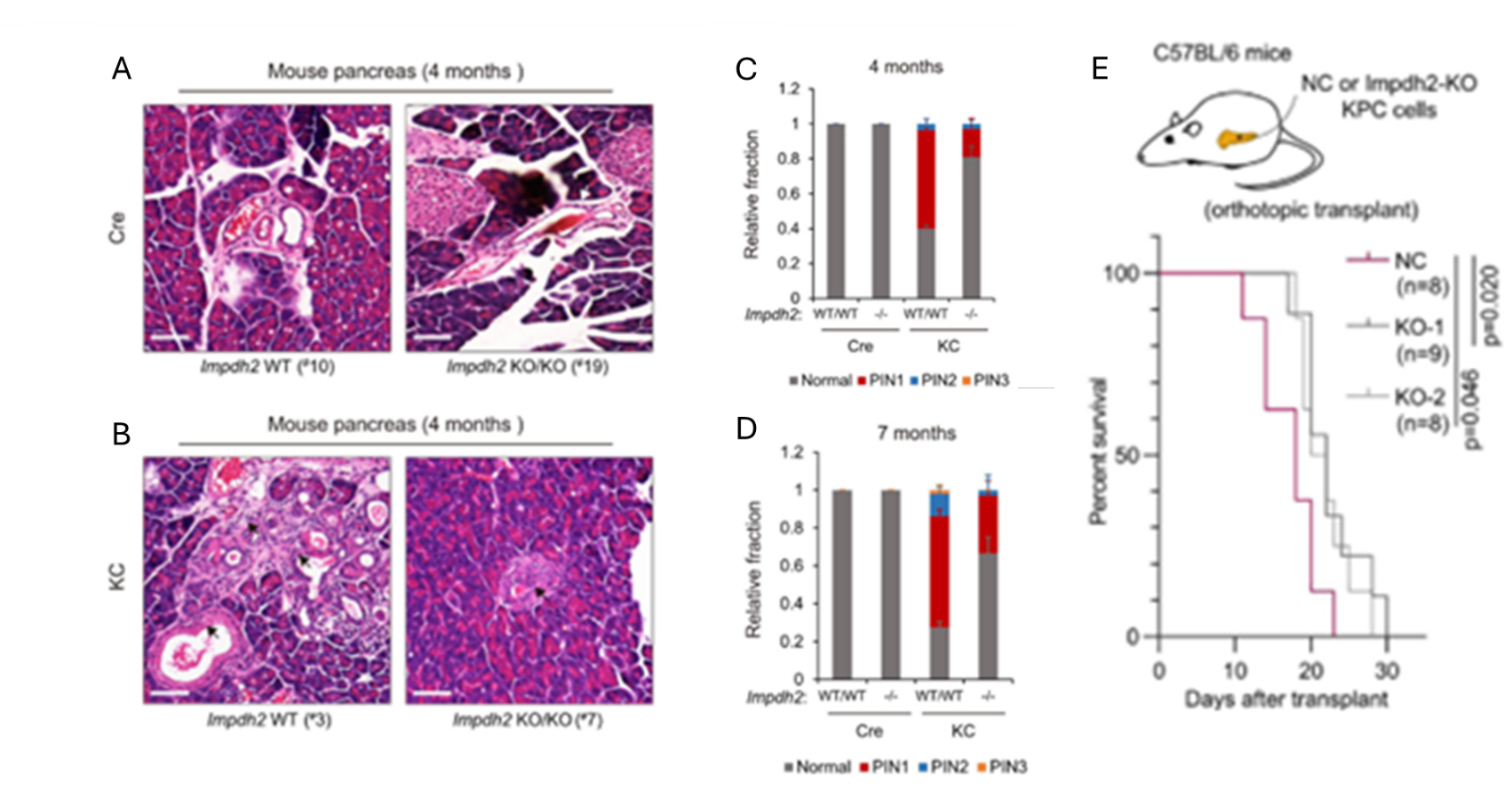
Figura 33. La inhibición de DNGB o la eliminación condicional de Impdh2 inhibe la carcinogénesis pancreática y la progresión del cáncer in vivo. Imágenes representativas de la tinción con H&E en muestras de tejido pancreático de ratones de control y (Cre; Impdh2-/-) (A), o de ratones KC y (KC; Impdh2-/-) (B). Frecuencia de los estadios de las lesiones de neoplasia intraepitelial pancreática (PanIN o PIN) (PIN 1, PIN 2 y PIN 3) en ratones de diferentes genotipos de 4 meses de edad (C) o de 7 meses de edad (D). Gráfico de supervivencia global de los ratones trasplantados ortotópicamente con células KPC de control negativo, una línea celular de PDAC transformada o células KPC con deleción de Impdh2 (E). Imagen reproducida de Wu et al., Gut, 2026, con licencia CC BY 4.0.
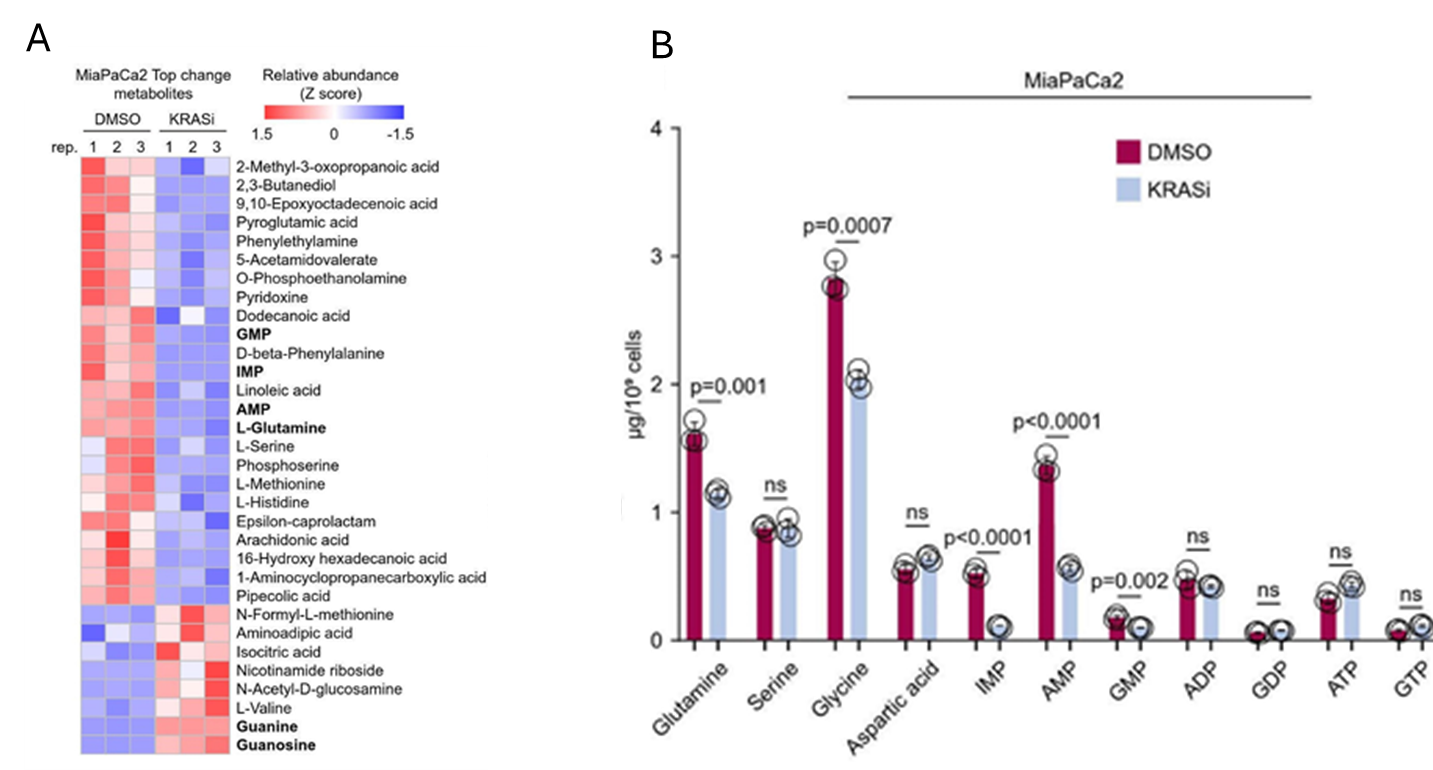
Figura 34. El KRAS mutante estimula la biosíntesis de novo de nucleótidos de adenina en mayor medida que la de nucleótidos de guanina. (A) Mapa de calor que muestra los metabolitos que experimentan cambios significativos y consistentes tras la inhibición aguda del KRAS mutante (G12C) en la línea celular transformada de PDAC, las células MiaPaCa2, según lo determinado mediante ensayos de metabolómica no dirigida. (B) Cuantificación de aminoácidos y nucleótidos de purina mediante LC-MS/MS dirigido en las células de control y con inhibición de KRAS (control: DMSO durante 120 min; inhibición de KRAS: 100 nM de AMG510 durante 120 min). Imagen reproducida de Wu et al., Gut, 2026, con licencia CC BY 4.0.
Conclusiones del estudio
- Antes de este estudio, la biosíntesis de novo de nucleótidos de guanina era una vulnerabilidad metabólica poco valorada en el PDAC con mutación en KRAS. La metabolómica desempeñó un papel importante al demostrar que KRAS potencia de forma específica la producción de nucleótidos de adenina, lo que provoca una escasez de nucleótidos de guanina y hace que las células del PDAC dependan de la IMPDH2, la enzima clave responsable de convertir el monofosfato de inosina en nucleótidos de guanina.
- Es importante destacar que, dado que IMPDH2 no está regulada por la vía de señalización canónica de KRAS, las células del PDAC no pueden compensar su inhibición mediante los mecanismos típicos de retroalimentación de dicha vía. Esto condujo al descubrimiento de que el bloqueo de IMPDH2 provoca un agotamiento irreversible de los nucleótidos de guanina, lo que altera la síntesis de ADN y ARN y frena el crecimiento tumoral.
- Estos hallazgos tienen implicaciones para las terapias futuras, ya que sugieren que la degradación selectiva de la IMPDH, en lugar de una inhibición generalizada, podría constituir una estrategia más precisa para el tratamiento del PDAC.
Microbioma
Aclaración de los mecanismos que relacionan la microbiota con la esclerosis múltiple
Introducción. Se ha demostrado que los metabolitos microbianos influyen en la regulación inmunitaria, la inflamación y la función neurológica, lo que sugiere que podrían contribuir a la actividad o la progresión de la enfermedad. Diversos estudios han identificado anomalías en la microbiota intestinal de pacientes con esclerosis múltiple (EM), pero aún no se ha establecido el papel que desempeñan los microbios intestinales y sus metabolitos en la patología de la EM.
Datos preliminares y objetivos del estudio. El objetivo de este estudio era determinar si determinados microbios intestinales y metabolitos fecales circulantes se asocian con el empeoramiento de la EM o con la transición a una forma progresiva de la enfermedad, mediante el análisis de datos clínicos, del microbioma y metabolómicos longitudinales de los participantes en una cohorte de EM bien caracterizada [19].
Métodos. En este estudio se analizó a los participantes de la cohorte del Estudio Longitudinal Integral de la Esclerosis Múltiple (CLIMB), junto con los datos clínicos asociados, las imágenes de resonancia magnética y las muestras de plasma y heces almacenadas. Los investigadores realizaron un seguimiento de los pacientes durante aproximadamente dos años y los clasificaron en grupos definidos por: 1) enfermedad estable, 2) empeoramiento de la discapacidad y 3) transición de la EM recidivante-remitente a la EM progresiva. Se realizó una secuenciación metagenómica de tipo «shotgun» en muestras de heces para caracterizar la microbiota intestinal. Tanto las muestras de heces como las de suero se analizaron mediante perfiles metabolómicos globales para medir los metabolitos microbianos y los derivados del huésped. Se utilizaron modelos estadísticos para identificar asociaciones entre los taxones microbianos, los niveles de metabolitos y los cambios en los resultados clínicos, incluyendo la progresión de la discapacidad, los hallazgos de la resonancia magnética y las medidas de calidad de vida.
Resultados. Se identificaron asociaciones significativas entre la composición de la microbiota intestinal, los perfiles de metabolitos y la progresión de la EM. Las personas que experimentaron un empeoramiento de la enfermedad o una transición a una EM progresiva mostraron niveles reducidos de varios taxones microbianos beneficiosos, incluidas especies conocidas por producir ácidos grasos de cadena corta y otros metabolitos antiinflamatorios (Figura 35). Por el contrario, los niveles más elevados de metabolitos producidos por estos microbios beneficiosos se asociaron con una enfermedad estable. Las características microbianas y los niveles de metabolitos se asociaron con la puntuación de discapacidad, los indicadores de actividad de la enfermedad en la resonancia magnética y los resultados comunicados por los pacientes, lo que indica que los cambios metabólicos derivados del microbioma pueden desempeñar un papel directo en la progresión de la EM (Figura 36).
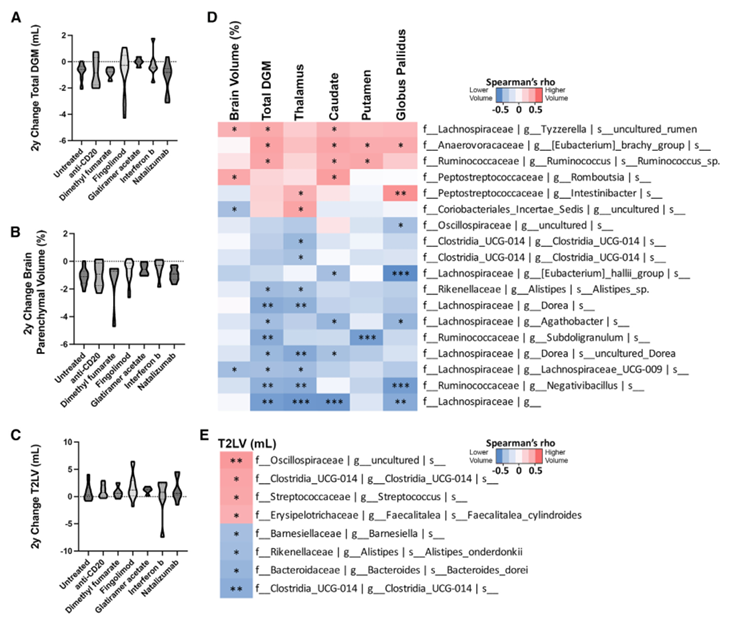
Figura 35. Los microbios intestinales se correlacionan con los cambios observados a lo largo de dos años en la resonancia magnética de 3T. Cambios en el DGM (A), el volumen parenquimatoso total del cerebro (B) y el T2LV estratificados según el DMT (C). Las correlaciones de Spearman muestran que los taxones microbianos se correlacionan de forma significativa y positiva (rojo) o negativa (azul) con los cambios observados a lo largo de dos años en las medidas volumétricas de 3T (D). La correlación de Spearman demuestra que los taxones se correlacionan positiva (rojo) o negativamente (azul) con el T2LV (E). Imagen reproducida de Schwerdtfeger et al., Cell Rep Med, 2025, con licencia CC BY 4.0.
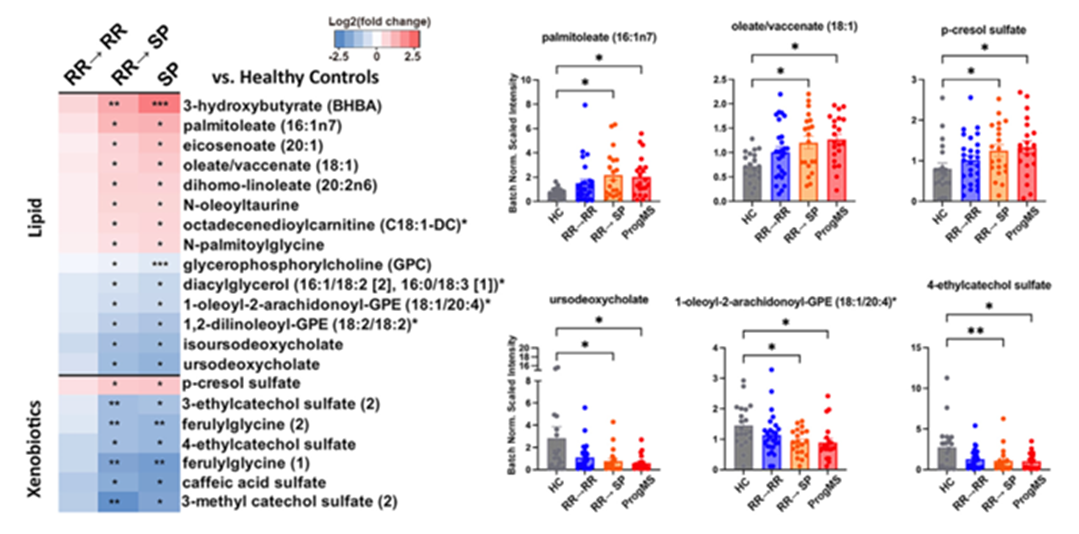
Figura 36. Metabolitos séricos relacionados con el desarrollo de la EM progresiva. (A) Todos los metabolitos séricos que presentaron alteraciones significativas en pacientes que pasaron de una enfermedad de tipo recidivante-remitente (RR) a una enfermedad progresiva estable (SP). (B) Gráficos de barras con los valores individuales de las alteraciones metabólicas biológicamente relevantes según el estado de la enfermedad. Imagen reproducida de Schwerdtfeger et al., Cell Rep Med, 2025, con licencia CC BY 4.0
Conclusiones del estudio
- Los resultados de este estudio respaldan la idea de que la microbiota intestinal podría influir en la esclerosis múltiple a través de vías de señalización metabólica y la modulación inmunitaria, y los taxones microbianos y metabolitos identificados podrían servir como biomarcadores de la progresión de la enfermedad o como nuevas dianas terapéuticas que podrían modularse mediante la dieta.
- Sin la metabolómica, los investigadores solo habrían podido observar cambios en las especies microbianas y no habrían podido determinar qué vías metabólicas se habían alterado, qué moléculas bioactivas podrían influir en la señalización inmunitaria y la neuroinflamación, ni qué moléculas podrían investigarse más a fondo como posibles dianas terapéuticas.
Comprender la influencia del microbioma en la fisiopatología de la diabetes tipo 2
Introducción. La diabetes tipo 2 (DT2) es una enfermedad metabólica compleja en la que influyen factores genéticos, ambientales y microbianos. Cada vez hay más pruebas que sugieren que la microbiota intestinal contribuye a la salud metabólica mediante la producción de metabolitos bioactivos que afectan a la fisiología del huésped, incluyendo el metabolismo de la glucosa y la inflamación. Aunque los estudios de secuenciación de la microbiota han identificado taxones microbianos asociados a la DT2, no explican por completo los mecanismos bioquímicos funcionales que vinculan a los microbios intestinales con la enfermedad metabólica.
Datos preliminares y objetivos del estudio. Dado que los metabolitos microbianos actúan como moléculas de señalización que influyen en el metabolismo del huésped, los investigadores trataron de caracterizar cómo la actividad metabólica microbiana contribuye al riesgo y la progresión de la diabetes tipo 2 mediante la secuenciación metagenómica y análisis metabolómicos globales [20].
Métodos. En este estudio se analizaron cohortes con estado diabético conocido y datos de fenotipado metabólico. Se utilizó la secuenciación metagenómica para caracterizar la composición y la abundancia de la microbiota intestinal. Se analizaron muestras de plasma y heces mediante perfiles metabolómicos globales para medir una amplia gama de metabolitos derivados del huésped y de los microbios. Se utilizaron análisis estadísticos para identificar metabolitos y taxones microbianos asociados con la DT2 y rasgos metabólicos relacionados, como la resistencia a la insulina y el control glucémico. Se utilizaron análisis integrativos para identificar conexiones entre microbios y metabolitos específicos con el fin de determinar cómo las vías metabólicas microbianas influyen en los fenotipos metabólicos del huésped.
Resultados. Las personas con DT2 presentaban perfiles de microbioma y metabolitos distintos en comparación con las personas sanas. Varios taxones microbianos se asociaron con concentraciones alteradas de metabolitos implicados en el metabolismo de los aminoácidos, el metabolismo de los lípidos y las vías de fermentación microbiana. La metabolómica reveló metabolitos específicos de origen microbiano que se correlacionaban significativamente con los indicadores de resistencia a la insulina y regulación de la glucosa (Figura 37). Curiosamente, se observó que los metabolitos asociados a la DT2 se revertían en respuesta a una intervención dietética o de ejercicio (Figura 38). Algunos metabolitos mostraron asociaciones más fuertes con el riesgo de DT2 que los propios taxones microbianos, lo que sugiere que la actividad metabólica microbiana, más que la simple abundancia microbiana, podría ser un factor clave de los cambios metabólicos relacionados con la enfermedad.
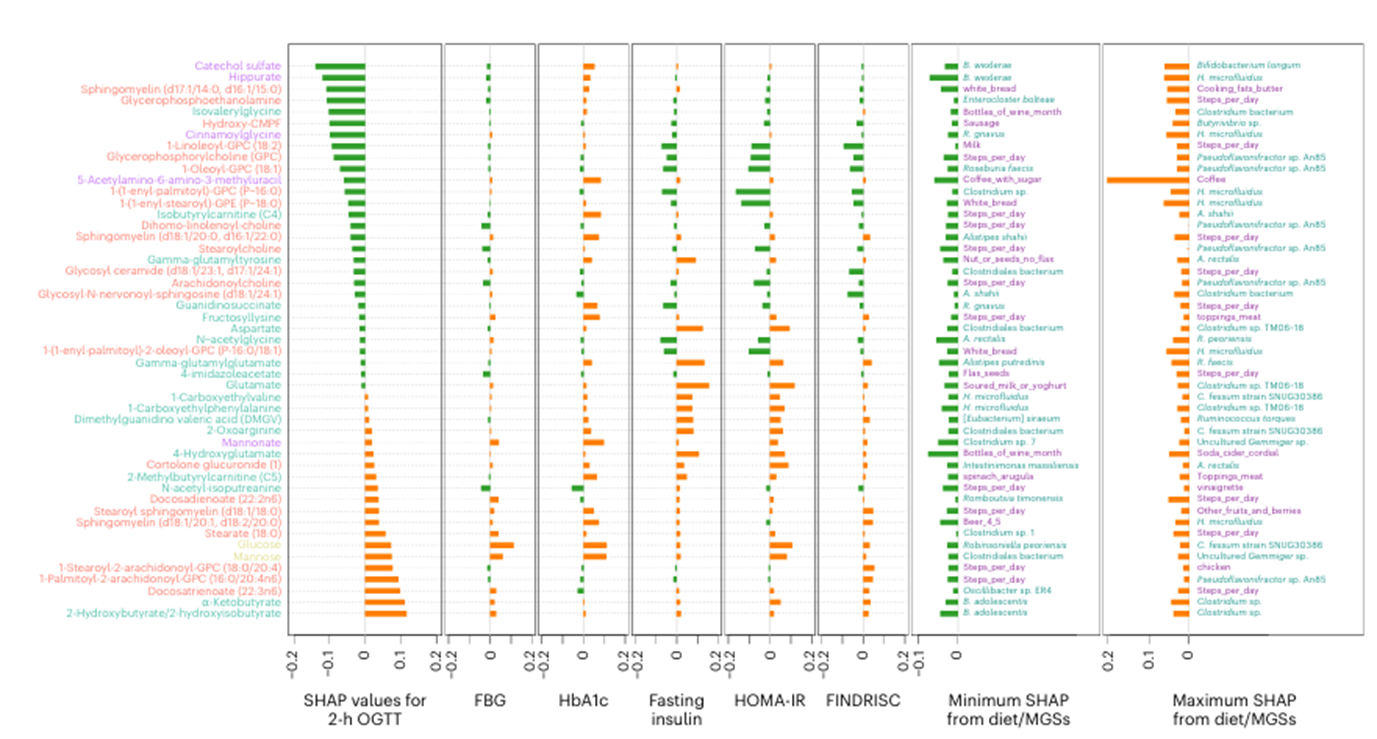
Figura 37. Características de la microbiota intestinal que explican la intolerancia a la glucosa. Los diez principales metabolitos identificados como características importantes en la prueba de tolerancia oral a la glucosa (OGTT) de dos horas, la glucemia en ayunas (FBG), la HbA1c, la insulina en ayunas, la evaluación del modelo homeostático de resistencia a la insulina (HOMA-IR) o la puntuación finlandesa de riesgo de diabetes (FINDRISC) (n = 49). Imagen reproducida de Wu et al., Nat Med, 2025, con licencia CC BY 4.0.
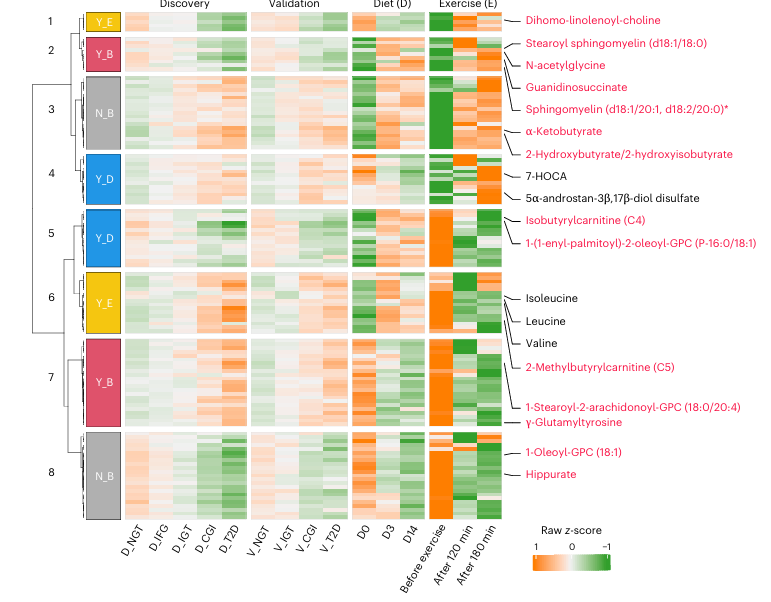
Figura 38. Respuestas de los metabolitos asociados a la prediabetes y la diabetes tipo 2 tras una intervención dietética de dos semanas o antes y después del ejercicio. Mapa de calor que muestra los metabolitos superpuestos implicados en el metabolismo de aminoácidos, lípidos y xenobióticos (n = 123) en dos ensayos clínicos de intervenciones con dieta (14 días) o ejercicio durante 1 hora (antes, 120 y 180 minutos después del ejercicio) con esos 502 metabolitos alterados en la prediabetes y la DT2. Las respuestas invertidas (Y, sí; N, no) por la dieta (D), el ejercicio (E) o ambos (B) se agruparon y se muestran en colores distintos junto a las ramas de agrupación de filas. Los metabolitos representativos están etiquetados en rojo, y otros cinco en negro. Imagen reproducida de Wu et al., Nat Med, 2025, con licencia CC BY 4.0.
Una relación poco estudiada entre los síntomas del síndrome del intestino irritable y las funciones cognitivas
Introducción. El síndrome del intestino irritable (SII) es un trastorno gastrointestinal caracterizado por dolor abdominal recurrente y alteraciones en los hábitos intestinales. Las interacciones entre el cerebro, el intestino y la microbiota se están reconociendo como importantes reguladores de la función gastrointestinal, la percepción de los síntomas y el estado de ánimo, lo que las ha convertido en objetivos para la intervención terapéutica en el SII. La terapia cognitivo-conductual (TCC) es una intervención eficaz dirigida al cerebro que enseña habilidades de procesamiento de la información para abordar los factores psicológicos que se sabe que exacerban los síntomas abdominales, como las estrategias de afrontamiento inadaptadas, la preocupación intensa y la reactividad al estrés.
Datos preliminares y objetivos del estudio. Un grupo de investigación demostró anteriormente, en un amplio ensayo clínico aleatorizado, que dos programas de TCC adaptados al SII resultaban eficaces para producir una mejora sostenida de los síntomas gastrointestinales, en comparación con un programa educativo sobre el SII que servía de control para los efectos inespecíficos derivados del hecho de recibir tratamiento. Basándose en estos hallazgos, el equipo del estudio planteó la hipótesis de que la TCC alivia los síntomas modulando principalmente el componente cerebral del eje cerebro-intestino-microbioma (BGM) y que las señales microbianas al cerebro en forma de metabolitos neuroactivos, incluidos los ácidos grasos de cadena corta y la serotonina, podrían modular la respuesta a los efectos biológicos de la TCC. Por lo tanto, el objetivo de este estudio fue determinar si las firmas metabólicas en pacientes con SII están asociadas con el rendimiento cognitivo y los síntomas relacionados con el BGM [21].
Métodos. Los pacientes elegibles fueron asignados aleatoriamente a recibir 10 sesiones de TCC en la consulta o 4 sesiones de TCC realizadas principalmente en el domicilio, con un contacto mínimo con el terapeuta, durante una fase aguda de 10 semanas. Todos los participantes se sometieron a sesiones de resonancia magnética (RM) al inicio del estudio y tras el tratamiento. Se utilizaron cuestionarios clínicos para evaluar la gravedad de los síntomas gastrointestinales. Las muestras de heces se analizaron mediante secuenciación del gen 16S rRNA y metabolómica no dirigida. Los análisis estadísticos identificaron metabolitos asociados con el estado del SII, el rendimiento cognitivo y la gravedad de los síntomas.
Resultados. De los 84 participantes con SII que se sometieron a pruebas de neuroimagen, 58 fueron clasificados como «respondedores a la TCC», mientras que 26 fueron clasificados como «no respondedores», basándose en una disminución de 50 puntos o más en la Escala de Gravedad de los Síntomas del SII tras el tratamiento. La beta-diversidad microbiana fue significativamente diferente entre los respondedores y los no respondedores, y la abundancia de serotonina —un neurotransmisor derivado del microbioma— aumentó significativamente en los respondedores. Tras la TCC, los que respondieron mostraron una disminución de la conectividad entre múltiples regiones asociadas a redes cerebrales específicas relacionadas con la regulación emocional (Figura 39). Cabe destacar que estos cambios se acompañaron de una disminución significativa tanto del dolor abdominal como del estrés percibido. Entre los pacientes que respondieron a la TCC, los niveles de Bacteroides eran más altos al inicio del estudio en comparación con los que no respondieron (Figura 40). Se utilizó un clasificador de bosque aleatorio que contenía 11 géneros bacterianos para predecir la respuesta a la TCC. Este clasificador fue capaz de predecir con precisión la respuesta a la TCC, lo que demuestra que el microbioma puede servir como un biomarcador eficaz para evaluar la respuesta potencial de un paciente al tratamiento y priorizar a los pacientes con probabilidades de responder positivamente a la TCC.
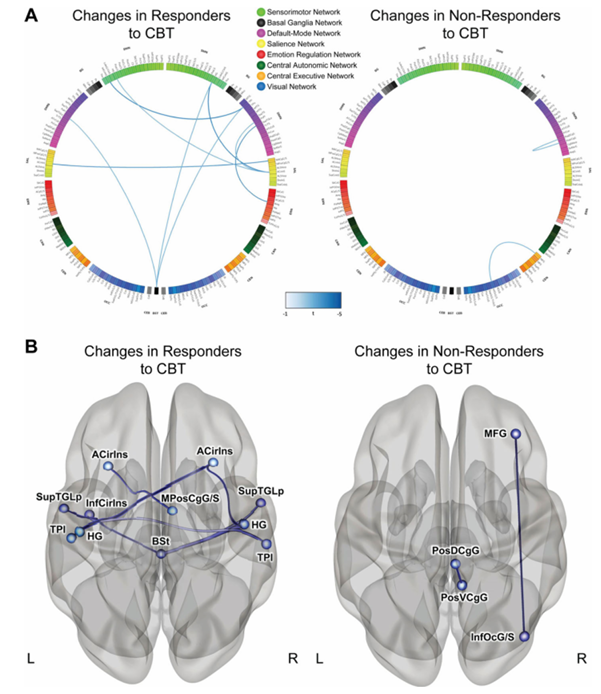
Figura 39. Cambios en la conectividad funcional entre los pacientes que respondieron y los que no respondieron a la TCC. (A) Connectogramas que muestran las diferencias de conectividad por pares entre los grupos. Las líneas azules indican disminuciones significativas en la conectividad. (B) Regiones que mostraron diferencias significativas entre los pacientes que respondieron y los que no respondieron a la TCC. Imagen reproducida de Jacobs et al., Microbiome, 2021, bajo licencia CC BY 4.0.
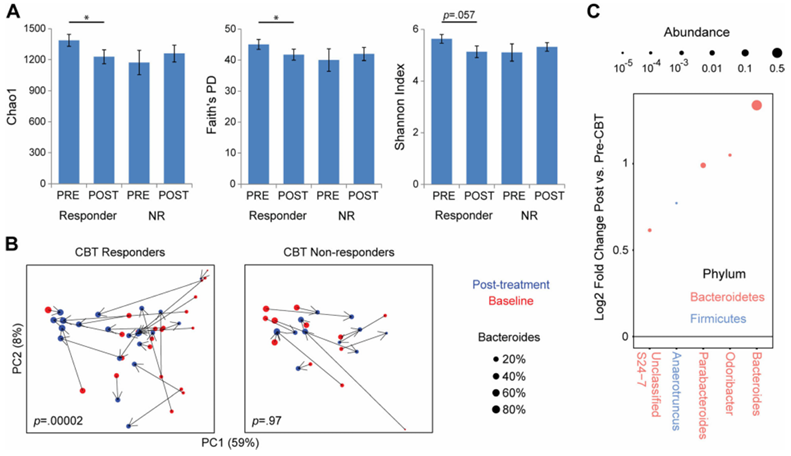
Figura 40. Las personas que responden a la TCC presentan una composición alterada de la microbiota intestinal tras la TCC, caracterizada por una expansión de Bacteroides. (A) Se muestra la diversidad alfa microbiana fecal de las personas que responden a la TCC y de las que no responden (NR) en la línea de base (PRE) y tras la TCC (POST). (B) Análisis de coordenadas principales de los datos de secuencias de ARN ribosómico 16S antes y después de la TCC, estratificados según el estado de respuesta a la TCC. Cada punto representa una muestra, coloreada según el momento temporal (rojo = inicio, azul = post-TCC) y cuyo tamaño depende de la abundancia de Bacteroides. (C) Se muestran los géneros microbianos con una asociación estadísticamente significativa con el estado de respuesta a la TCC. Imagen reproducida de Jacobs et al., Microbiome, 2021, con licencia CC BY 4.0.
Conclusiones del estudio
- Estos hallazgos apuntan a un mecanismo relacionado con la microbiota por el cual los pacientes con SII experimentan dolor sin que exista una patología subyacente, y abren nuevas vías para el uso de componentes del eje BGM como biomarcadores en el tratamiento del SII.
- La metabolómica fue fundamental para este estudio, ya que permitió medir directamente las moléculas de señalización entre el intestino y el cerebro que relacionan la disfunción gastrointestinal con los síntomas cognitivos en el síndrome del intestino irritable, lo que contribuyó a desentrañar la compleja relación entre la microbiota y la salud humana.
Salud de la población
Identificación de nuevas relaciones entre las vías bioquímicas y las enfermedades no transmisibles para explicar la multimorbilidad
Introducción. La multimorbilidad, es decir, la coexistencia de múltiples enfermedades crónicas, constituye una importante carga sanitaria a nivel mundial cuya prevalencia va en aumento. Antes de poder abordar adecuadamente este problema, debemos comprender mejor los factores de riesgo de las enfermedades y los mecanismos de su progresión. Aunque muchas enfermedades crónicas comparten factores de riesgo y mecanismos biológicos, la mayoría de los estudios se limitan a una sola enfermedad. El perfil molecular podría identificar vías biológicas comunes que predisponen a las personas a padecer múltiples enfermedades. Con este fin, los investigadores realizaron un perfil metabolómico no dirigido en muestras de plasma recogidas de la cohorte Norfolk del Estudio Prospectivo Europeo sobre el Cáncer (EPIC) [22].
Datos preliminares y objetivos del estudio. El objetivo de este estudio era identificar las vías metabólicas comunes, determinar qué asociaciones están determinadas por factores de riesgo modificables y destacar los metabolitos relacionados con el desarrollo de múltiples enfermedades crónicas.
Métodos. Se analizaron las muestras de plasma de 11 966 personas de la cohorte EPIC-Norfolk mediante metabolómica no dirigida. Las enfermedades incidentes y la mortalidad se identificaron mediante un seguimiento a largo plazo utilizando historias clínicas electrónicas vinculadas, datos de hospitalización y registros de cáncer que abarcaban más de 219 000 personas-año. Las asociaciones entre los niveles de metabolitos y la incidencia de enfermedades se evaluaron mediante modelos de riesgos proporcionales de Cox ajustados por edad y sexo. Se utilizó la regresión lineal para examinar las asociaciones de los metabolitos con la multimorbilidad.
Resultados. Los hallazgos revelaron 458 metabolitos asociados con al menos un resultado clínico, lo que dio lugar a un total de 1226 asociaciones entre metabolitos y enfermedades. El 65,5 % de los metabolitos significativos se asociaron con dos o más enfermedades, lo que indica un amplio solapamiento biológico entre las afecciones crónicas. Se observaron fuertes conexiones entre las enfermedades cardiometabólicas, respiratorias, renales y hepáticas, y muchos metabolitos mostraron direcciones de asociación consistentes en múltiples afecciones (Figura 41). Los análisis de mediación mostraron que muchas relaciones entre metabolitos y enfermedades estaban asociadas a factores de riesgo comunes, como la obesidad, el tabaquismo, la inflamación, la resistencia a la glucosa y los niveles elevados de lípidos. Algunos metabolitos se asociaron de forma exclusiva con enfermedades específicas, mientras que 30 metabolitos se relacionaron de manera significativa con el desarrollo de multimorbilidad (Figura 42).
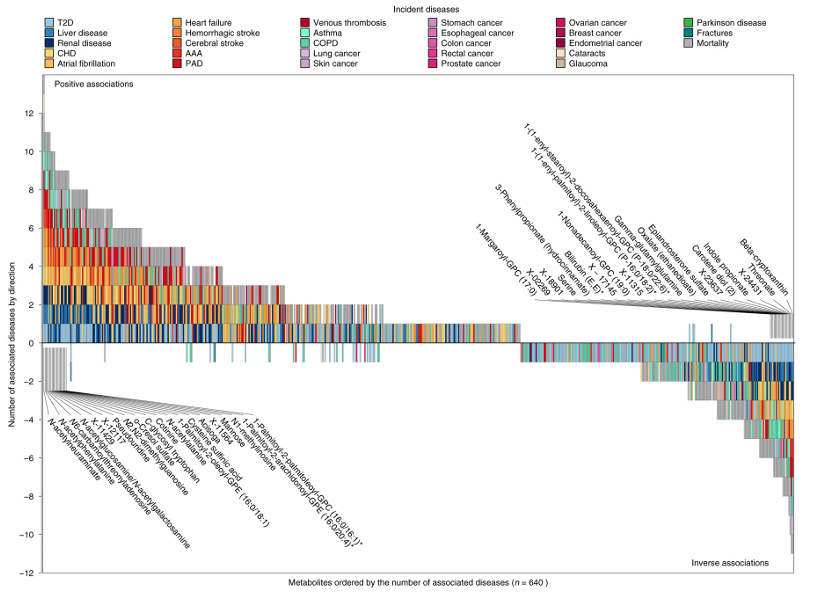
Figura 41. Gráfico de ladrillos que muestra la clasificación de los metabolitos en función del número de resultados clínicos asociados. Se consideró significativo un valor de p < 0,001, teniendo en cuenta las 28 enfermedades analizadas para cada metabolito. El eje x muestra la clasificación de cada metabolito según el número de resultados clínicos asociados, contando las asociaciones inversas como números negativos para simplificar la representación de los resultados. El eje Y muestra el número de metabolitos asociados, donde los números positivos indican asociaciones positivas y los números negativos indican asociaciones inversas. Los colores de cada casilla indican el criterio de valoración de la enfermedad asociado. Se han anotado los metabolitos seleccionados con múltiples criterios de valoración asociados. Imagen reproducida de Pietzner et al., Nat Med, 2021, con licencia CC BY 4.0.
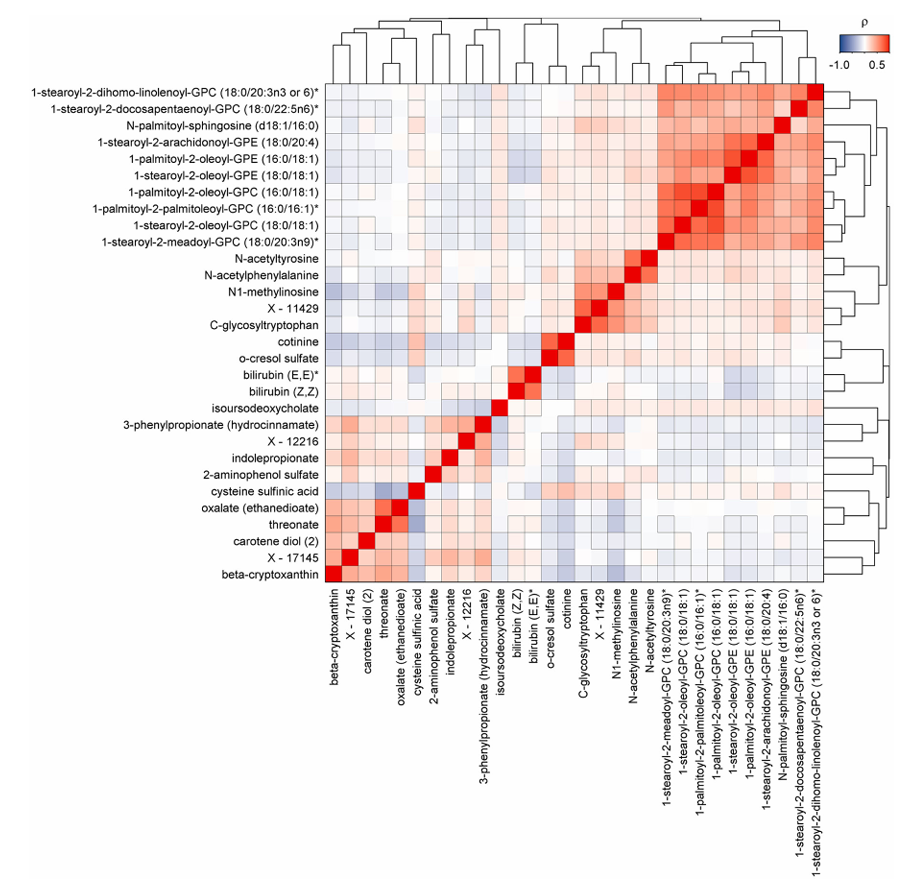
Figura 42. Mapa de calor de correlaciones por pares de metabolitos candidatos a la multimorbilidad. Matriz de correlaciones por pares de metabolitos plasmáticos asociados de forma significativa con la incidencia de multimorbilidad por enfermedades no transmisibles. Los colores indican correlaciones positivas (rojo) o inversas (azul), y los recuadros negros indican significación estadística tras la corrección por pruebas múltiples. Imagen reproducida de Pietzner et al., Nat Med, 2021, bajo licencia CC BY 4.0.
Conclusiones del estudio
- En este estudio, la metabolómica no dirigida resultó fundamental para identificar asociaciones entre las vías bioquímicas comunes a múltiples enfermedades, lo que proporcionó tanto información sobre los mecanismos que subyacen al riesgo de enfermedad como conocimientos prácticos para el desarrollo de futuras terapias.
- En muchos casos, las vías que se asociaron con más de una enfermedad mostraron las mismas tendencias de asociación con cada una de ellas por separado. Esto sugiere que las futuras intervenciones podrían centrarse en las vías comunes para prevenir múltiples enfermedades.
- Los resultados de este estudio también podrían servir de guía para identificar subtipos de multimorbilidad, al analizar cómo esas vías se relacionan con la coexistencia de enfermedades aparentemente no relacionadas entre sí.
Predicción de la edad biológica para evaluar mejor el riesgo de enfermedad
Introducción. La edad cronológica es un factor de riesgo importante para numerosas enfermedades, pero no refleja el complejo proceso de envejecimiento biológico ni la considerable variabilidad del envejecimiento biológico entre individuos. La diferencia entre el envejecimiento cronológico y el envejecimiento biológico podría ser un indicador más revelador del estado de salud que la edad cronológica por sí sola. El envejecimiento biológico refleja interacciones complejas entre factores genéticos, de estilo de vida y ambientales, y la identificación de biomarcadores que representen mejor este proceso podría mejorar la comprensión del riesgo de enfermedad y las trayectorias de salud. La metabolómica es un enfoque prometedor, ya que los metabolitos circulantes reflejan tanto la actividad metabólica endógena como las exposiciones externas, incluyendo la dieta, los medicamentos y los factores ambientales.
Datos preliminares y objetivos del estudio. Los estudios previos sobre la predicción de la edad metabólica se han visto limitados por el reducido tamaño de las muestras, los paneles de metabolitos específicos o los rangos de edad restringidos, lo que puede reducir la precisión predictiva y la generalizabilidad. Por lo tanto, los objetivos de este estudio fueron desarrollar un modelo metabolómico robusto para la predicción de la edad mediante la realización de perfiles metabolómicos no específicos en INTERVAL, una gran cohorte basada en la población, y evaluar si las estimaciones de la edad metabólica resultantes se asociaban con resultados de salud [23].
Métodos. El estudio INTERVAL es un estudio de cohorte prospectivo con aproximadamente 50 000 participantes, integrado en una muestra aleatoria de donantes de sangre. En este estudio, se analizaron muestras de plasma de 12 000 donantes de sangre sanos, de entre 18 y 75 años, mediante metabolómica global. Se utilizó la regresión de cresta combinada con el método bootstrapping para construir y validar internamente modelos que predijeran la edad cronológica a partir de los perfiles de metabolitos. También se construyeron modelos independientes utilizando únicamente metabolitos endógenos y datos específicos por sexo para hombres y mujeres. Las predicciones de edad metabolómica resultantes se comprobaron posteriormente en el estudio Netherlands Epidemiology of Obesity (NEO) para determinar si las diferencias entre la edad metabolómica prevista y la edad cronológica se asociaban con características de salud.
Resultados. Los modelos metabolómicos de predicción de la edad demostraron un buen rendimiento, ya que explicaban más del 80 % de la variación en la edad cronológica cuando se incluían tanto metabolitos endógenos como xenobióticos (Figura 43). Entre los metabolitos clave que contribuyeron de manera significativa a la predicción de la edad se encontraban la hidroxiasparagina, el vanililmandelato y la 5,6-dihidrouridina. Cuando se aplicó el modelo a la cohorte NEO, una edad metabolómica superior a la edad cronológica se asoció con la obesidad y las enfermedades cardiovasculares, en parte debido a que los metabolitos xenobióticos reflejaban el uso de medicación y factores ambientales como la cotinina (Figura 44). El estudio también identificó 163 metabolitos que diferían entre hombres y mujeres, y los modelos específicos por sexo mostraron un alto rendimiento predictivo, pero solo una correlación moderada entre sí, lo que sugiere que estos modelos captaron diferencias significativas entre sexos en los patrones metabólicos relacionados con la edad.
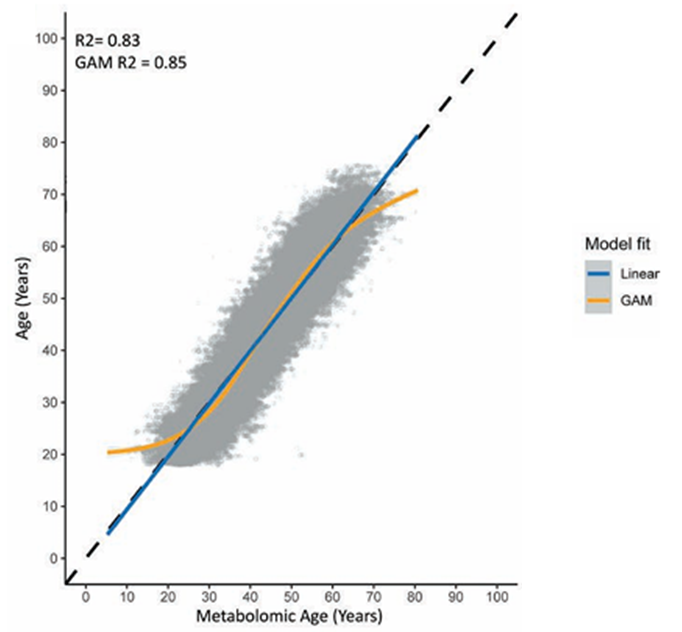
Figura 43. Gráfico de correlación entre la edad metabolómica (edad prevista) en el eje x y la edad cronológica en el eje y para todos los metabolitos endógenos y xenobióticos analizados. Imagen reproducida de Faquih et al., J Gerontol A Biol Sci Med Sci, 2025, bajo licencia CC BY 4.0.
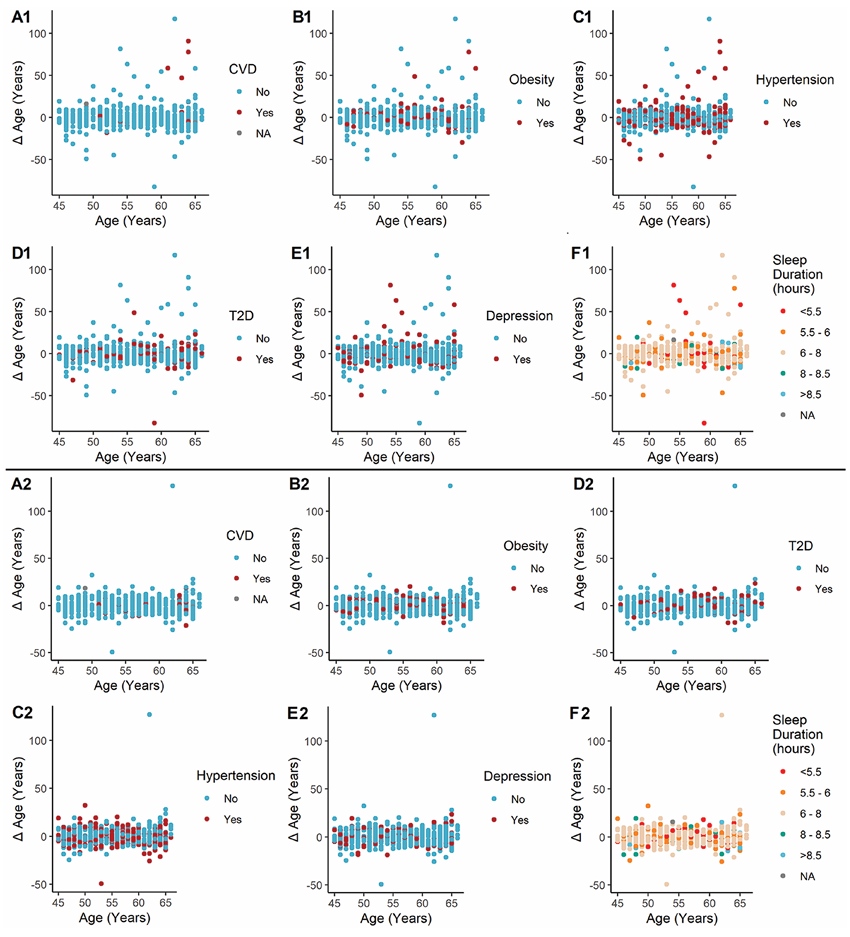
Figura 44. Gráficos de dispersión que muestran la diferencia de edad (Δ edad) según las predicciones del modelo de metabolitos endógenos + xenobióticos (A1-F1) y del modelo de solo metabolitos endógenos (A2-F2). NEO = Estudio epidemiológico sobre la obesidad en los Países Bajos. Imagen reproducida de Faquih et al., J Gerontol A Biol Sci Med Sci, 2025, con licencia CC BY 4.0.
Conclusiones del estudio
- La incorporación de una amplia gama de metabolitos endógenos y xenobióticos, medidos mediante metabolómica global, dio lugar a un modelo metabolómico de edad sólido que superó en eficacia a muchos enfoques anteriores.
- Estos resultados demuestran que los modelos de predicción de la edad metabolómica tienen potencial como herramientas para estudiar la biología del envejecimiento e identificar a las personas con mayor riesgo de padecer enfermedades relacionadas con la edad.
Relación entre las variantes patógenas heterocigotas y el curso de la enfermedad
Introducción. Aunque los estudios de asociación de todo el genoma (GWAS) de variantes comunes han identificado muchos loci relacionados con los niveles de metabolitos, estas variantes suelen tener efectos modestos y no indican directamente la función del gen. Las variantes raras perjudiciales identificadas mediante la secuenciación del exoma completo pueden aportar una visión funcional más sólida, ya que alteran de manera sustancial la actividad de las proteínas. Sin embargo, la mayoría de las variantes raras se dan en estado heterocigoto, y sus consecuencias fisiológicas no se conocen bien.
Datos preliminares y objetivos del estudio. En este estudio, los investigadores se propusieron integrar la metabolómica con la secuenciación del exoma para determinar de forma sistemática cómo las variantes heterocigotas raras influyen en los niveles de metabolitos y en los rasgos humanos, así como para determinar si estas variantes pueden revelar efectos graduales sobre la función génica y las vías metabólicas [24].
Métodos. Se utilizaron la metabolómica no dirigida y la secuenciación del exoma completo para identificar asociaciones entre los metabolitos y las variantes raras perjudiciales. Se midieron los niveles plasmáticos y urinarios de una amplia gama de metabolitos en grandes cohortes poblacionales, entre ellas el UK Biobank (UKB) y la German Chronic Kidney Disease (GCKD). Se realizaron pruebas de agregación de variantes raras para evaluar si los grupos de variantes raras y potencialmente dañinas dentro de un gen se asociaban con los niveles de metabolitos. Se utilizaron simulaciones computacionales con modelos de redes metabólicas a escala genómica para validar los resultados.
Resultados. Se identificaron 235 asociaciones significativas entre genes y metabolitos relacionadas con variantes raras perjudiciales en múltiples vías metabólicas, muchas de las cuales no se habían descrito anteriormente. Estas asociaciones pusieron de relieve genes que codifican enzimas y transportadores que influyen en las concentraciones de metabolitos en plasma o orina. Varias variantes genéticas con diferentes impactos funcionales previstos produjeron cambios graduales en los niveles de metabolitos, lo que proporcionó evidencia de efectos dependientes de la dosis sobre la función metabólica (Figura 45). Cabe destacar que las variantes raras en genes transportadores de sulfato, como SCL13A1 y SLC26A1, se asociaron fuertemente con las concentraciones de sulfato circulante y también se relacionaron con la estatura y los rasgos musculoesqueléticos (Figura 46). La integración de la evidencia genética con modelos metabólicos computacionales respaldó la relevancia funcional de muchas asociaciones.
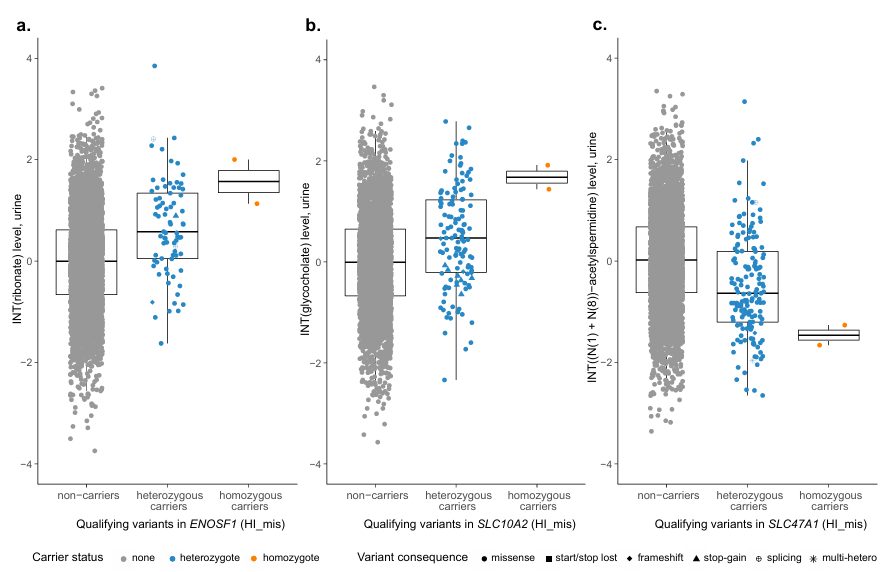
Figura 45. Niveles de metabolitos según el estado de portador de variantes relevantes para los genes significativamente asociados con más de un portador homocigótico. En el eje Y se muestran los niveles de metabolitos en orina tras la transformación normal inversa y el ajuste por covariables, mientras que en el eje X se representan los no portadores y los portadores heterocigóticos de variantes relevantes en la máscara HI_mis. El color y la forma de los símbolos indican el estado de portador de una variante y su consecuencia, respectivamente. Los portadores de múltiples variantes heterocigotas que cumplen los criterios se indican con un asterisco. Las cajas abarcan desde el percentil 25 hasta el 75 de los niveles de metabolitos, la mediana se indica mediante una línea y las barras terminan en el último valor observado dentro de 1,5*(rango intercuartílico) de distancia de la caja. Los niveles medianos de ribonato (n = 4618) (A), glicocolato (n = 3753) (B) y (N(1) + (N(8))-acetilspermidina (n = 4619) (C) son más extremos en los portadores homocigotos que en los heterocigotos, lo que refleja un efecto dosis-respuesta. Imagen reproducida de Scherer et al., Nat Genet, 2025, con licencia CC BY 4.0.
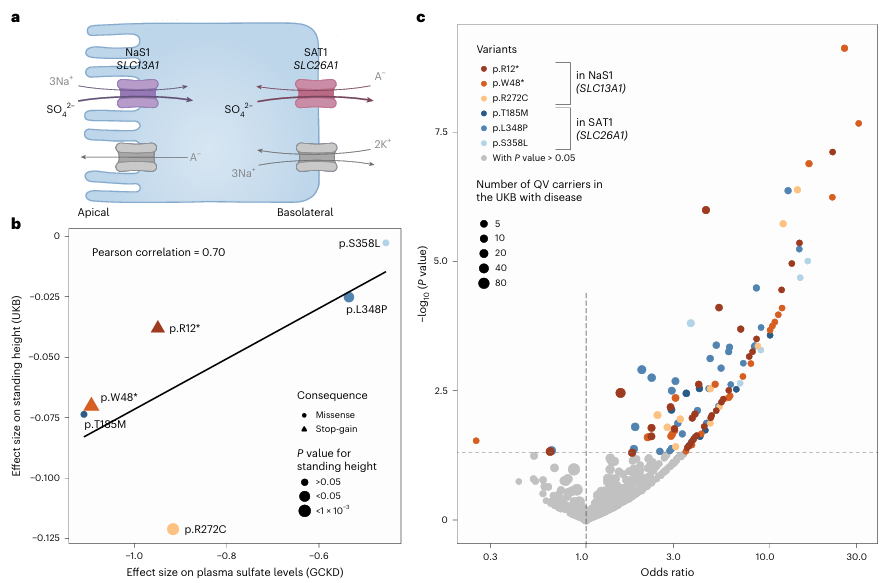
Figura 46. El impacto de las variantes funcionales de importancia en los genes SLC13A1 y SLC26A1 sobre la estatura, los rasgos musculoesqueléticos y las fracturas respalda el papel del sulfato plasmático como indicador intermedio. (A) Representación esquemática del mecanismo de reabsorción de sulfato en el que intervienen NaS1, codificado por SLC13A1 en la membrana apical, y SAT1, codificado por SLC26A1 en la membrana basolateral de las células epiteliales. (B) El diagrama de dispersión muestra la relación entre los tamaños del efecto de seis variantes cualificantes sobre los niveles de sulfato plasmático en la cohorte GCKD (eje x) y sobre la estatura de pie en la cohorte UKB (eje y). (C) Gráfico de volcán que muestra las odds ratio (eje x) y -log10 (valores p) (eje y) para las asociaciones de las seis variantes cualificadas con enfermedades musculoesqueléticas y fracturas en el UKB. Imagen reproducida de Scherer et al., Nat Genet, 2025, con licencia CC BY 4.0.
Conclusiones del estudio
- La metabolómica fue fundamental para demostrar que incluso las variantes perjudiciales heterocigotas pueden provocar cambios metabólicos cuantificables.
- Los efectos fenotípicos graduales que se observaron muestran cómo la variación genética determina el metabolismo y los rasgos humanos.
- En general, los resultados de este estudio ponen de manifiesto el valor de la metabolómica a la hora de obtener una lectura funcional que permita interpretar variantes genéticas raras, y muestran cómo la combinación de perfiles metabólicos a gran escala con la secuenciación puede aportar nuevos conocimientos sobre la compleja relación entre la variación genética y la enfermedad.
Matrices de muestra alternativas
Leche materna
Introducción. Comprender el metaboloma de la leche materna puede aportar información útil sobre la nutrición y la salud del lactante; sin embargo, la gran variabilidad de la composición de la leche entre personas y en una misma persona a lo largo del tiempo dificulta la definición de concentraciones estándar para muchos componentes. Estudios metabolómicos previos han analizado la variación en la composición de la leche en función de las etapas de la lactancia, el estado de salud de la madre y las poblaciones, pero son relativamente pocos los que se han centrado en identificar los metabolitos fundamentales que están presentes de forma constante en madres de distintos orígenes.
Datos preliminares y objetivos del estudio. El objetivo de este estudio era caracterizar el metaboloma de la leche materna en mujeres lactantes sanas de diversos orígenes, con el fin de definir la leche materna de manera uniforme entre las madres lactantes e identificar o corroborar qué elementos son fundamentales para la nutrición, el crecimiento y el desarrollo del lactante [25].
Métodos. Se recogieron muestras de leche materna de 31 mujeres que representaban diversos orígenes raciales, étnicos y alimentarios. Las participantes también completaron un cuestionario sobre salud y estilo de vida. Las muestras de leche se analizaron mediante perfiles metabolómicos globales y se estudiaron las relaciones entre los perfiles metabólicos y diversas características maternas o del lactante, como la edad materna, el IMC, el número de partos, la edad del lactante y factores relacionados con el estilo de vida. Las vías metabólicas asociadas a las variables clave se identificaron mediante un análisis de enriquecimiento.
Resultados. Se detectaron 389 metabolitos en todas las muestras. En general, la abundancia de estos metabolitos comunes varió considerablemente entre las participantes del estudio. Al agruparlos por vías biológicas, los xenobióticos mostraron la mayor variabilidad en toda la población del estudio, mientras que los nucleótidos fueron los menos variables. La edad del lactante, la edad materna, el número de partos vivos previos y el IMC previo al embarazo se asociaron con diferencias en el metaboloma de la leche (Figura 47). Los lípidos esteroles y los carbohidratos variaban con la edad del lactante, ya que el colesterol y el sulfato de colesterol aumentaban a medida que los lactantes crecían. La edad materna se asoció con cambios en los compuestos orgánicos de oxígeno, y el número de nacidos vivos se relacionó con la variación en los ácidos orgánicos, los ácidos nucleicos, los carbohidratos y los lípidos.
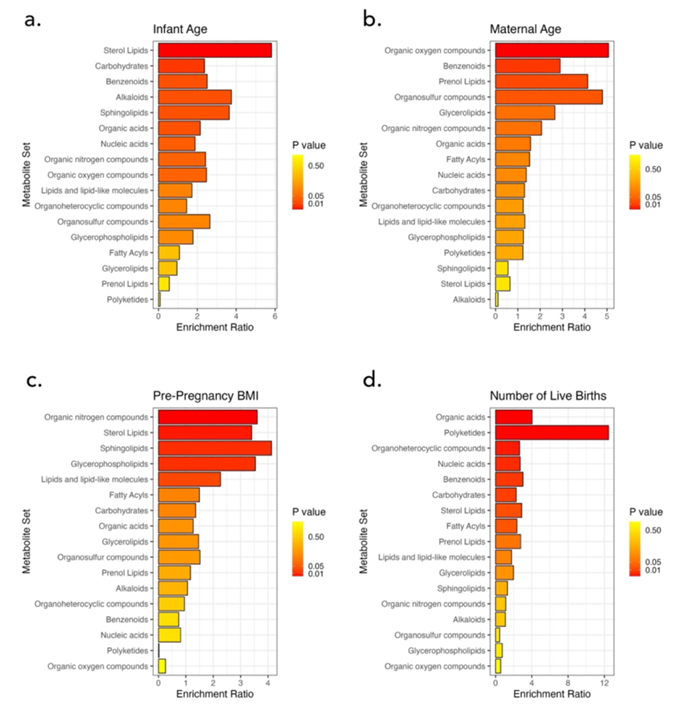
Figura 47. Análisis cuantitativo de enriquecimiento de conjuntos de metabolitos (MSEA) según (A) la edad del bebé, (B) la edad de la madre, (C) el número de nacidos vivos y (D) el IMC previo al embarazo. Imagen reproducida de Holmes et al., Sci Rep, 2024, con licencia CC BY 4.0.
Conclusiones del estudio
- Este estudio sugiere que la leche materna contiene un conjunto básico de metabolitos conservados que probablemente desempeñan un papel esencial en la nutrición y el desarrollo psicológico del lactante.
- Los metabolitos implicados en funciones celulares fundamentales, como los nucleótidos, la lactosa, la creatinina y el glutamato, mostraron la menor variabilidad, lo que sugiere que las moléculas que contribuyen a procesos críticos, como el metabolismo energético, son las que están reguladas con mayor rigor.
- Este estudio es otro ejemplo que demuestra cómo la metabolómica puede aportar conocimientos científicos sobre matrices de muestras poco habituales.
Humor acuoso
Introducción. El humor acuoso (HA) contiene valiosa información molecular sobre las enfermedades de la retina y puede obtenerse de forma no invasiva, lo que lo convierte en una fuente práctica para el descubrimiento de biomarcadores y el estudio de los mecanismos de las enfermedades. En estudios previos se han utilizado la metabolómica y la proteómica para investigar enfermedades oculares como el glaucoma, la degeneración macular neovascular relacionada con la edad (DMNE) y el edema macular diabético (EMD). Sin embargo, la interpretación de los datos moleculares obtenidos del HA puede verse sesgada por factores de confusión, como las variaciones en la concentración total de proteínas y el estado del cristalino (fáquico [el ojo contiene un cristalino natural] frente a pseudofáquico [el cristalino natural sustituido por una lente artificial para tratar las cataratas]).
Datos preliminares y objetivos del estudio. El objetivo de este estudio era determinar cómo influyen estos factores de confusión oculares en los perfiles proteómicos y metabolómicos de la hemorragia retiniana (HR), utilizando muestras de pacientes con DMAE neovascular y edema macular diabético (EMD), con el objetivo a largo plazo de mejorar la fiabilidad del descubrimiento de biomarcadores basados en la HR [26].
Métodos. Este estudio clínico prospectivo y transversal incluyó a 102 participantes con DMAE neovascular (nAMD), 18 con edema macular diabético (DME) y 18 pacientes con cataratas sin enfermedad retiniana como grupo de control. Se obtuvieron muestras de humor acuoso (HA) mediante paracentesis de la cámara anterior y se congelaron para su análisis. Se utilizó la plataforma de ensayo de extensión de proximidad Olink Target 96 para medir proteínas relacionadas con procesos inflamatorios, metabólicos, neurológicos y cardiovasculares. La concentración total de proteínas en el HA se midió por separado. Los metabolitos se analizaron utilizando Global Discovery Panel Metabolon. Se utilizaron modelos lineales con corrección por pruebas múltiples para identificar proteínas y metabolitos con abundancia diferencial entre los grupos, teniendo en cuenta al mismo tiempo posibles factores de confusión oculares, como el estado del cristalino, la concentración total de proteínas y la exposición a fármacos dilatadores de la pupila. Se utilizaron análisis de correlación, agrupamiento de perfiles proteicos y enriquecimiento de conjuntos de genes para identificar las vías biológicas asociadas a los patrones moleculares observados.
Resultados. El humor acuoso (HA) de los pacientes con DMAE neovascular (nAMD) y edema macular diabético (DME) contenía concentraciones totales de proteínas significativamente más altas que las de los controles. Los ojos pseudofáquicos (con lente artificial) presentaban concentraciones de proteínas en el HA más elevadas que los ojos fáquicos (con lente natural) y mostraban niveles aumentados de proteínas relacionadas con la remodelación de la matriz extracelular y las vías de señalización (Figura 48). Tras ajustar los resultados para tener en cuenta los factores de confusión, el número de proteínas diferenciales entre los grupos de pacientes y los controles disminuyó sustancialmente, lo que permitió identificar firmas proteicas asociadas a la enfermedad de mayor relevancia biológica. Se identificaron grupos de proteínas relacionados con la señalización neuronal, los péptidos antimicrobianos, las enzimas metabólicas y las respuestas al estrés oxidativo, que mostraron un enriquecimiento de proteínas previamente asociadas a la degeneración macular relacionada con la edad (nAMD) y al EMD. Los perfiles metabolómicos se vieron menos afectados por estos factores de confusión. El análisis metabolómico condujo al descubrimiento inesperado de que las gotas dilatadoras de la pupila que contienen fenilefrina/tropicamida se asociaban con niveles elevados de varias proteínas inflamatorias, en particular la interleucina-6 (IL-6), probablemente debido a la presencia del metabolito de la fenilefrina 3-hidroximandelato en la HA (Figura 49). La metabolómica también proporcionó información sobre los fenotipos de la enfermedad. Por ejemplo, los niveles elevados de glucosa y fructosa y la reducción de 1,5-anhidroglucitol en las muestras de DME indicaban un mal control glucémico e hiperglucemia crónica.
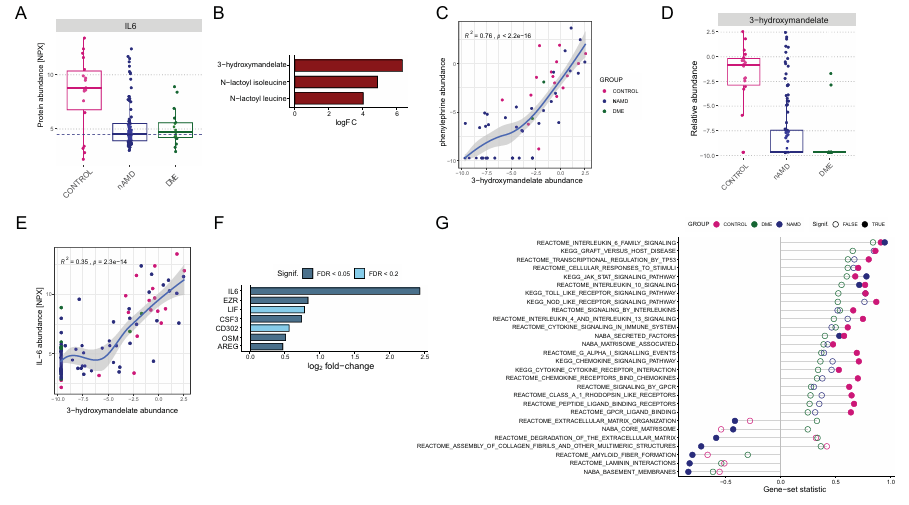
Figura 48. Asociación entre la exposición a la fenilefrina/3-hidroximandelato y los perfiles proteicos de la AH. (A) Los diagramas de caja muestran los valores de abundancia proteica relativa (NPX) de la IL-6 en los tres grupos de pacientes. La línea punteada representa el umbral utilizado para dividir el grupo de control con vistas al posterior análisis de abundancia diferencial. (B) Gráfico de barras de los metabolitos significativamente asociados con niveles altos frente a bajos de IL-6 dentro del grupo de control (FDR < 0,05). (C) Correlación entre los niveles de 3-hidroximandelato y fenilefrina. (D) Los diagramas de caja muestran los valores de abundancia relativa de metabolitos para el 3-hidroximandelato en los tres grupos de pacientes. (E) Igual que en (C), pero para la correlación entre los niveles de 3-hidroximandelato e IL-6. (F) Gráfico de barras de las proteínas asociadas a niveles altos frente a bajos de 3-hidroximandelato en el grupo de nAMD, con los cambios logarítmicos (log2) indicados y los niveles de significación representados por la escala de colores. (G) Se muestran los resultados del GSEA para los niveles altos frente a los bajos de 3-hidroximandelato en el grupo de nAMD. Imagen reproducida de Titz et al., Transl Vis Sci Technol, 2024, con licencia CC BY 4.0.

Figura 49. Influencia del estado del cristalino en los perfiles proteicos del humor acuoso. (A) Mapa de calor que muestra la abundancia diferencial de proteínas, ajustada en función de la concentración proteica, entre los ojos pseudofáquicos y fáquicos dentro de los grupos de pacientes con DMAE neovascular y EMD. Los cambios logarítmicos (log2) se visualizan mediante un degradado de color, y las proteínas con diferencias estadísticamente significativas se marcan (*P < 0,05, ajustado por FDR). El mapa de calor se limita a las 15 proteínas principales clasificadas según el valor absoluto de logFC. (B) Se presentan los resultados del GSEA para el estado del cristalino (pseudofáquico frente a fáquico). Imagen reproducida de Titz et al., Transl Vis Sci Technol, 2024, con licencia CC BY 4.0.
Conclusiones del estudio
- Los resultados de este estudio muestran que el análisis proteómico de la AH puede verse muy influido por factores de confusión oculares y relacionados con el procedimiento, mientras que la metabolómica se ve menos afectada por ellos.
- En este estudio, la metabolómica identificó los colirios dilatadores de la pupila que contienen fenilefrina como un factor de confusión hasta ahora desconocido. El 3-hidroximandelato presente en estos colirios se asoció con un aumento de los niveles de proteínas inflamatorias. Esto no solo ha permitido descubrir un nuevo mecanismo en la biología ocular, sino que también pone de manifiesto que, en futuros estudios, será necesario tener en cuenta la exposición a la fenilefrina para interpretar con precisión los datos ómicos del humor acuoso.
- Este estudio también sirve como ejemplo de cómo la metabolómica puede aportar conocimientos científicos fundamentales a partir de una matriz de muestra poco habitual.
Frotis cervicales
Introducción. La microbiota vaginal desempeña un papel fundamental en la salud reproductiva de la mujer. Los entornos vaginales saludables suelen estar dominados por especies de Lactobacillus, en particular Lactobacillus crispatus, que se asocian con efectos protectores, mientras que las comunidades microbianas más diversas y ricas en anaerobios se relacionan con la vaginosis bacteriana (VB). Lactobacillus iners desempeña un papel paradójico en la VB, ya que, a diferencia de otras especies de Lactobacillus, se asocia con peores resultados clínicos y un mayor riesgo de desarrollar VB, a pesar de ser un organismo dominante en la mayoría de las mujeres. El tratamiento estándar de la VB con metronidazol a menudo da lugar a comunidades dominadas por L. iners, que son inestables y propensas a la recaída. Abordar este reto clínico requiere nuevas estrategias que puedan modular el microbioma vaginal para dar lugar al crecimiento de una microbiota duradera y asociada a la salud.
Datos preliminares y objetivos del estudio. La biología de L. iners es poco conocida, principalmente porque resulta difícil cultivarla y estudiarla in vitro. En comparación con otras especies de Lactobacillus, tiene un genoma más pequeño y una capacidad metabólica limitada, lo que sugiere que depende de nutrientes externos. A partir de estos hallazgos previos, el objetivo de este estudio fue identificar las dependencias metabólicas clave de L. iners y determinar si esta vulnerabilidad podría aprovecharse para modificar la microbiota vaginal hacia composiciones más beneficiosas y mejorar los resultados del tratamiento de la VB [27].
Métodos. Se obtuvieron cepas clínicas de una amplia cohorte de mujeres sudafricanas y estadounidenses que incluía tanto casos de VB como casos sin VB. A partir de estas cepas se elaboró un extenso catálogo genómico que permitió comparar las capacidades metabólicas entre las distintas especies microbianas. Las muestras de lavado cervicovaginal se analizaron mediante metabolómica global y se determinó el perfil de la composición de la microbiota mediante la secuenciación del gen del ARN ribosómico 16S. Se utilizaron análisis estadísticos para correlacionar los niveles de metabolitos con la estructura de la comunidad bacteriana (por ejemplo, comunidades dominadas por Lactobacillus frente a comunidades asociadas a la VB).
Resultados. Los hallazgos revelaron que el crecimiento de L. iners depende exclusivamente de la cisteína. Se observó un crecimiento robusto de diversas cepas de L. iners en medios de cultivo suplementados con L-cistina, mientras que no se produjo crecimiento alguno en cultivos con bajos niveles de L-cistina. Se demostró que L. iners tiene una capacidad limitada para utilizar fuentes complejas de cisteína, lo que explica su requisito específico de crecimiento y pone de relieve una vulnerabilidad metabólica clave. En las muestras clínicas, los niveles de cisteína fueron significativamente más altos en mujeres sin vaginosis bacteriana (VB) y se correlacionaron positivamente con la abundancia de Lactobacillus, mientras que se correlacionaron negativamente con las bacterias asociadas a la VB (Figura 50). Los análisis genómicos revelaron que L. iners carece de muchos de los sistemas de transporte de cisteína y moléculas que contienen cisteína presentes en otras especies de Lactobacillus, lo que sugiere que tiene una flexibilidad metabólica limitada. Los inhibidores de la captación de cistina suprimieron selectivamente el crecimiento de L. iners sin afectar significativamente a otras especies de Lactobacillus, y en condiciones de competencia
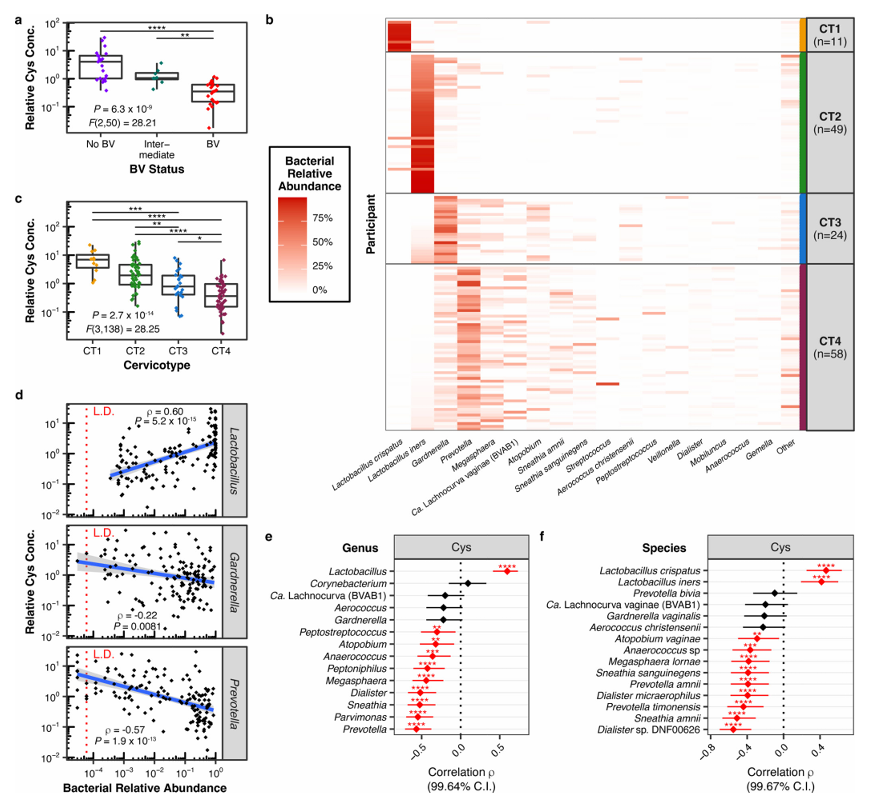
Figura 50. Las concentraciones de cistina (Cys) vaginal son más elevadas en las mujeres sin VB y se correlacionan con una microbiota dominada por Lactobacillus. (A) Concentración relativa de Cys según el estado de VB en el líquido de lavado cervicovaginal (CVL) de 53 mujeres sudafricanas (21 sin VB, 24 con VB y 8 en estado intermedio). (B) Composición de la microbiota bacteriana del tracto genital femenino (FGT) en 142 mujeres sudafricanas no infectadas por el VIH (incluidas las 53 de A), determinada mediante secuenciación del gen 16S rRNA bacteriano. (C) Concentraciones relativas de Cys por CT en el líquido cervical (CTL) de las mujeres de (B). (D) Correlación de rangos de Spearman de dos colas entre las concentraciones de Cys y las abundancias relativas de las bacterias de los géneros Lactobacillus, Gardnerella y Prevotella. (E, F) Coeficientes de correlación de Spearman de dos colas (ρ) con intervalos de confianza ajustados entre las concentraciones de Cys y las abundancias relativas de cada género (E) o especie (F) detectadas con una prevalencia >50 % en la cohorte (n = 142). Imagen reproducida de Bloom et al., Nat Microbiol, 2022, con licencia CC BY 4.0.
Conclusiones del estudio
- Este estudio identificó la dependencia de la cisteína como una característica biológica definitoria de L. iners, lo que ofrece una explicación de las dificultades asociadas al cultivo de esta especie.
- L. iners presenta una limitación única en su capacidad para transportar y utilizar diversas fuentes de cisteína, lo que la hace especialmente dependiente de formas específicas de cisteína exógena. Esta limitación metabólica ayuda a explicar su comportamiento ecológico y la distingue de especies más relacionadas con la salud, como L. crispatus.
- Los resultados del estudio también indican que los niveles de cisteína procedentes del huésped podrían influir en que prevalezcan las comunidades dominadas por Lactobacillus o las asociadas a la vaginosis bacteriana, lo que sugiere que la disponibilidad de metabolitos podría ser un factor determinante de la estructura de la comunidad microbiana en el entorno vaginal.
- En general, este estudio ha demostrado la viabilidad del concepto de que los inhibidores de la captación de cistina pueden suprimir activamente la L. iners y potenciar la eficacia del metronidazol, lo que sugiere un nuevo enfoque para el tratamiento de la vaginosis bacteriana.
Líquido sinovial
Introducción. La osteoartritis afecta a millones de personas y contribuye de manera significativa a la discapacidad. La osteoartritis postraumática (OPT) del tobillo reviste especial importancia, ya que suele afectar a personas más jóvenes y puede desarrollarse años después de la lesión inicial. A pesar de su prevalencia, los procesos biológicos tempranos que impulsan la OPT siguen sin conocerse bien, y los métodos diagnósticos habituales —basados en pruebas de imagen y exámenes clínicos— no detectan los cambios tempranos de la enfermedad, lo que limita las oportunidades de intervención precoz. La lesión articular aguda desencadena respuestas inflamatorias en el líquido sinovial, incluyendo niveles elevados de citocinas, metaloproteinasas de la matriz y daño celular, todo lo cual puede contribuir a la degradación del cartílago y a la degeneración articular a largo plazo. El líquido sinovial actúa como un depósito de metabolitos derivados de los tejidos articulares, y estudios previos sugieren que la composición lipídica cambia con la lesión, la inflamación y la artritis. Sin embargo, dado que los trabajos anteriores se basaban en técnicas analíticas relativamente limitadas, las alteraciones metabólicas completas que se producen tras la lesión siguen sin conocerse bien.
Datos preliminares y objetivos del estudio. El objetivo de este estudio era utilizar la metabolómica global para identificar los cambios metabólicos relacionados con los lípidos asociados a lesiones agudas y a los procesos tempranos de la enfermedad, con el objetivo a largo plazo de descubrir biomarcadores y posibles dianas terapéuticas que pudieran mejorar la comprensión y el tratamiento de la PTOA [28].
Métodos. El estudio consistió en un análisis de cohorte retrospectivo de pacientes con fracturas intraarticulares unilaterales de tobillo que requirieron cirugía. Se incluyó a veinte pacientes, y se extrajo líquido sinovial (LS) tanto del tobillo lesionado como del tobillo contralateral no lesionado en el momento de la intervención quirúrgica. A un subgrupo de siete pacientes también se le realizó una segunda extracción bilateral de LS seis meses después. Las concentraciones de metabolitos se corrigieron para tener en cuenta la dilución que se produjo durante la recogida por lavado, utilizando un método de normalización basado en la urea que comparaba los niveles de urea en el líquido sinovial con los del suero. Tras el perfil metabólico global, se utilizó la clasificación de bosque aleatorio para identificar los metabolitos clave que diferenciaban el LS del tobillo lesionado del LS de control. Se realizaron análisis de correlación para identificar las conexiones biológicas entre los metabolitos lipídicos, las citocinas inflamatorias y las metaloproteinasas de la matriz.
Resultados. Los ácidos grasos de cadena larga, los ácidos grasos poliinsaturados (PUFA), las esfingomielinas y los lisolípidos aumentaron de forma significativa en los tobillos fracturados en comparación con los controles contralaterales. El análisis de bosque aleatorio confirmó que los ácidos grasos y los lisolípidos eran los principales factores que distinguían las articulaciones lesionadas de las no lesionadas (Figura 51). Con el tiempo, muchas de estas anomalías lipídicas mostraron una resolución parcial, ya que varios ácidos grasos y esfingomielinas disminuyeron significativamente a los 6 meses de la cirugía. En consecuencia, la capacidad de los perfiles metabolómicos para distinguir las articulaciones lesionadas de las de control disminuyó a los 6 meses, lo que sugiere que la respuesta metabólica aguda a la lesión remite con el tiempo. Los metabolitos lipídicos mostraron fuertes correlaciones positivas con las citocinas inflamatorias y las metaloproteinasas de la matriz, lo que vincula la desregulación lipídica con los procesos de degradación tisular (Figura 52). La magnitud y el alcance de los cambios lipídicos se asociaron con la gravedad de la lesión, y las fracturas más graves mostraron mayores elevaciones de ácidos grasos, lisolípidos y metabolitos relacionados.
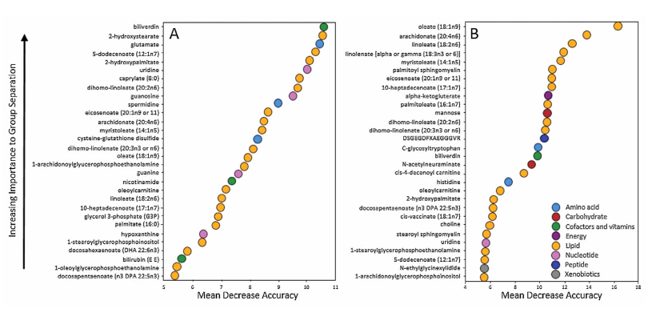
Figura 51. Análisis de clasificación mediante Random Forest de «Lesionados» frente a «Control contralateral» en la línea de base (A), y de la línea de base de «Lesionados» frente a «Lesionados a los 6 meses» (B). Imagen reproducida de Leimer et al., J Orthop Res, 2017, con licencia CC BY 4.0.
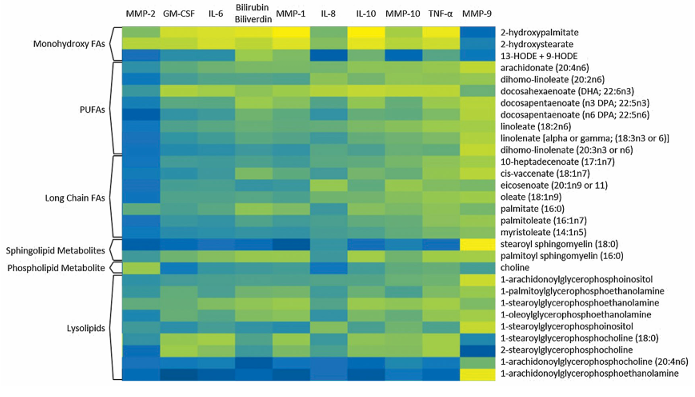
Figura 52. Correlaciones entre los metabolitos lipídicos y los niveles de citocinas en la misma cohorte de pacientes. Los compartimentos del mapa de calor van del negro al azul y al amarillo, donde el negro representa las correlaciones más débiles y el amarillo las más fuertes. Todos los valores de correlación significativos fueron positivos. Imagen reproducida de Leimer et al., J Orthop Res, 2017, bajo licencia CC BY 4.0.
Conclusiones del estudio
- Los resultados de este estudio sugieren que una fractura intraarticular aguda de tobillo produce un perfil lipídico característico en el líquido sinovial, marcado por un aumento de los ácidos grasos libres, los lisolípidos y los esfingolípidos.
- Las fuertes correlaciones entre los metabolitos lipídicos y las citocinas o las metaloproteinasas de la matriz sugieren que estas alteraciones lipídicas están estrechamente relacionadas con la inflamación, el daño tisular y la degradación del cartílago en la articulación lesionada.
- Dado que muchos de los cambios lipídicos provocados por la lesión fueron transitorios y se normalizaron a los seis meses de la intervención quirúrgica, la respuesta metabólica podría formar parte de un proceso agudo de lesión y cicatrización, y los aumentos tempranos de estos metabolitos también podrían activar vías de señalización que contribuyan a la degeneración articular a largo plazo y a la progresión hacia la artroplastia total de rodilla.
- Los metabolitos lipídicos pueden servir como biomarcadores de lesiones articulares tempranas y del riesgo de artrosis primitiva, así como posibles dianas para la intervención.
Conclusiones del capítulo
- La metabolómica puede aplicarse a numerosos diseños de estudio y a una amplia variedad de temas en la investigación científica básica.
- En la mayoría de los casos, la metabolómica resulta fundamental para obtener una perspectiva clave o realizar un descubrimiento crucial que impulse la investigación y la eleve a un nivel superior de relevancia.

Descarga la guía completa en formato PDF
Descargue esta completa guía desarrollada para enseñarle los entresijos de una de las herramientas ómicas más potentes de la caja de herramientas de cualquier científico: la metabolómica.
Descargar ahoraPóngase en contacto con nosotros
Hable con un experto
Solicite un presupuesto para nuestros servicios, obtenga más información sobre tipos de muestras y procedimientos de manipulación, solicite una carta de apoyo o envíe una pregunta sobre cómo la metabolómica puede hacer avanzar su investigación.
Sede social
617 Davis Drive, Suite 100
Morrisville, NC 27560
Dirección postal:
P.O. Box 110407
Research Triangle Park, NC 27709
+1 (919) 572-1711
Sede internacional
Metabolon GmbH
Zeppelinstraße 3
85399 Hallbergmoos
Alemania