Chapitre 5
La métabolomique au service de la recherche fondamentale
Dans ce chapitre, vous découvrirez comment la métabolomique permet de mettre en lumière des mécanismes qui échappent aux autres sciences omiques, afin de révolutionner les futures stratégies thérapeutiques ; comment elle a permis de mettre en évidence des informations clés qui ont considérablement fait progresser notre compréhension et les implications cliniques de l'axe microbiome-cerveau et de la santé des populations ; et comment elle peut générer des données pertinentes à partir de diverses matrices d'échantillons qui ne font généralement pas l'objet d'analyses.
Vue d'ensemble
La métabolomique a joué un rôle déterminant dans l'approfondissement de notre compréhension des principes scientifiques fondamentaux, grâce aux connaissances approfondies qu'elle apporte sur les mécanismes ayant un impact direct sur le phénotype et l'évolution naturelle des maladies, ainsi qu'à sa capacité à stimuler la découverte et à générer des hypothèses. Ces dernières années, elle a également apporté une contribution significative à des domaines d'intérêt particulier, notamment la recherche axée sur le microbiome et la santé des populations. Enfin, sa compatibilité avec des matrices d'échantillons inhabituelles la rend largement applicable à la plupart des domaines des sciences de la vie. Nous mettons ici en avant quelques études qui montrent comment la métabolomique peut élucider des mécanismes et être appliquée à d'autres types d'échantillons.
Mécanisme
Le remodelage cardiaque dans l'insuffisance cardiaque
Introduction. L'insuffisance cardiaque (IC) s'accompagne d'une altération de la flexibilité métabolique, qui empêche le cœur d'augmenter son métabolisme du glucose en réponse à un stress, ce qui entraîne une diminution de l'efficacité cardiaque et une progression de l'IC. Dans certaines circonstances, la fonction systolique du ventricule gauche (VG) peut être rétablie s'il reste du myocarde viable et si la cause sous-jacente de l'IC est traitée avec succès. La thérapie de resynchronisation cardiaque (TRC) est un traitement de l'IC dans lequel un bloc de branche gauche étendu entraîne une contraction désynchronisée entre le septum et la paroi latérale du VG. La CRT améliore rapidement l'hémodynamique cardiaque et l'efficacité de l'oxygénation, et une amélioration supplémentaire de la fonction ventriculaire gauche est observée à mesure que le remodelage se produit au fil du temps. Les mécanismes qui relient la CRT au remodelage cardiaque structurel à long terme restent flous. Il a été démontré que la CRT modifie le métabolome circulant au fil du temps, mais on ne sait pas clairement s'il s'agit d'une cause ou d'un effet.
Données préliminaires et objectifs de l'étude. En fin de compte, l'amélioration de la fonction contractile nécessite un apport accru en ATP au niveau de l'appareil contractile. Il est donc plausible que le degré de flexibilité métabolique conservé par le cœur défaillant soit déterminant pour sa capacité à se remodeler. L'objectif de cette étude était de tester cette hypothèse en caractérisant le degré de remodelage ventriculaire gauche en réponse à 6 mois de CRT et de déterminer si la CRT peut modifier de manière aiguë l'absorption des substrats, s'éloignant du phénotype métabolique de l'insuffisance cardiaque pour favoriser en fin de compte le remodelage [16].
Méthodes. Cette étude a analysé des patients atteints d’insuffisance cardiaque (IC) ainsi que des groupes témoins appropriés à l’aide d’un phénotypage clinique et d’un profilage métabolomique global. Après un jeûne nocturne, les patients ont été stabilisés sous perfusion d’insuline et de glucose pendant au moins une heure avant l’implantation du dispositif de resynchronisation cardiaque (CRT). Des mesures de la courbe pression-volume ont été effectuées et des échantillons artério-veineux coronariens ont été prélevés. La perfusion d'insuline/glucose a été interrompue et une perfusion d'intralipide a été mise en place pendant 15 minutes, après quoi les mesures ont été répétées. Les échantillons de sang artériel et veineux ont été analysés par métabolomique non ciblée, et la flexibilité métabolique a été évaluée en mesurant l'absorption de certains métabolites et en calculant l'efficacité de l'oxygénation myocardique. Des analyses statistiques et des analyses des voies métaboliques ont été utilisées pour identifier les métabolites associés à une altération de la flexibilité métabolique.
Résultats. La CRT a entraîné une augmentation significative de l'absorption des acides gras non estérifiés (AGNE), sans augmentation significative de l'absorption du glucose, du bêta-hydroxybutyrate ou du lactate (Figure 29). Une forte corrélation a été observée entre l'amélioration aiguë des performances hémodynamiques cardiaques induite par le raccourcissement du QRSd résultant de la CRT. Il existait une forte corrélation positive entre l'augmentation du travail systolique et l'absorption des AGNE. La modification de l'absorption des substrats en réponse à la CRT était corrélée au remodelage inverse à long terme du volume télédiastolique ventriculaire gauche (Figure 30). Une corrélation significative a été observée entre l'augmentation de l'absorption des NEFA et la réduction du VTDG. L'analyse lipidomique a montré que ce phénomène était dû à une augmentation des acides gras à chaîne longue et à chaîne moyenne.
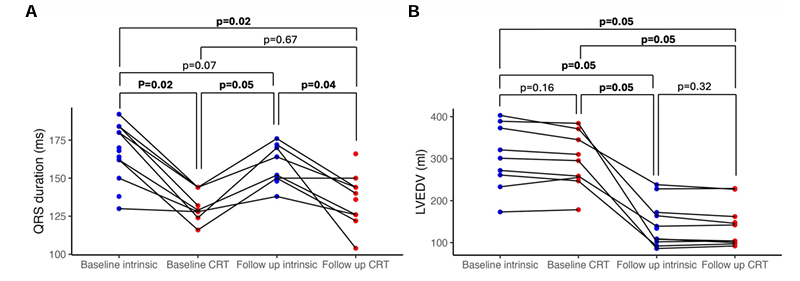
Figure 29. Remodelage ventriculaire gauche chronique après 6 mois de CRT, indépendamment des variations aiguës de la durée du QRS. Durée du QRS (A, 13 patients) et volume télédiastolique ventriculaire gauche (B, 9 patients) au moment de l'implantation initiale et lors du suivi à 6 mois, avec rythme intrinsèque et stimulation optimisée par thérapie de resynchronisation cardiaque. Image reproduite à partir de Green et al., Eur Heart J, 2025, sous licence CC BY 4.0.
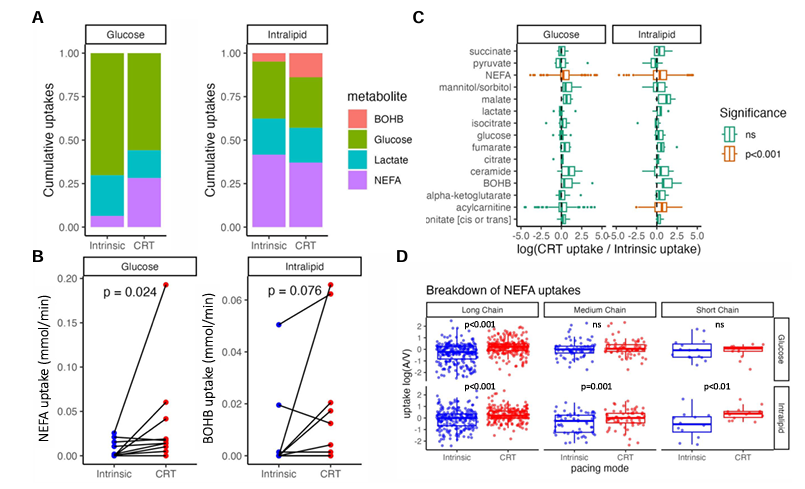
Figure 30. Effet de la CRT sur l'absorption des substrats cardiaques pendant des perfusions d'intralipide et d'insuline/glucose. (A) Proportion cumulative de l'absorption des acides gras libres (AGL), du bêta-hydroxybutyrate, du lactate et du glucose pendant la conduction intrinsèque et la CRT optimisée, sous perfusions d'intralipide et d'insuline/glucose. (B) Illustration de l'effet de la thérapie de resynchronisation cardiaque sur l'absorption des acides gras non estérifiés et du β-hydroxybutyrate lors de perfusions d'insuline/glucose et d'intralipides respectivement. (C) Analyse métabolomique illustrant la différence d'absorption des métabolites intermédiaires induite par le démarrage de la thérapie de resynchronisation cardiaque, où les valeurs positives indiquent une augmentation de l'absorption avec la thérapie de resynchronisation cardiaque. (D) Analyse lipidomique illustrant l'effet de la thérapie de resynchronisation cardiaque sur les absorptions [définies comme log (artériel/veineux)] d'acides gras non estérifiés de différentes longueurs de chaîne sous perfusion d'insuline/glucose et d'intralipid. Image reproduite à partir de Li et al., J Exp Med, 2014 sous licence CC BY 4.0
Conclusions de l'étude
- Ces résultats montrent que, dans la cardiomyopathie non ischémique, le cœur conserve une grande souplesse métabolique, et que la résynchronisation cardiaque permet ainsi de faire évoluer le phénotype métabolique de l'insuffisance cardiaque vers un phénotype plus physiologique caractérisé par l'absorption des acides gras libres.
- Le degré de flexibilité métabolique conservée est corrélé au remodelage inverse à long terme du volume télédiastolique ventriculaire gauche.
- Dans cette étude, la métabolomique a révélé que le remodelage cardiaque induit par la thérapie de resynchronisation cardiaque (CRT) pourrait résulter davantage des modifications du métabolisme cellulaire provoquées par cette thérapie que des effets aigus du raccourcissement du complexe QRS.
Identification d'une nouvelle cible thérapeutique pour la néphropathie diabétique
Introduction. La néphropathie diabétique (ND) est l'une des principales causes d'insuffisance rénale chronique et d'insuffisance rénale. Bien que les inhibiteurs du cotransporteur sodium-glucose de type 2 (SGLT2) aient été initialement développés pour réduire la glycémie en augmentant l'excrétion urinaire de glucose, des essais cliniques ont montré qu'ils offraient également une protection rénale et cardiovasculaire importante, même chez les patients non diabétiques. Cependant, les mécanismes biologiques responsables de ces effets protecteurs restent flous, notamment parce que le SGLT2 n'est exprimé que dans les cellules tubulaires proximales (CTP) du rein, ce qui soulève la question de savoir comment le ciblage d'un transporteur dans une population cellulaire limitée peut améliorer la fonction rénale globale.
Données préliminaires et objectifs de l'étude. Des études antérieures suggèrent que les reins des patients diabétiques présentent des altérations métaboliques importantes, notamment une glycolyse accrue et un dysfonctionnement mitochondrial, que l'inhibition du SGLT2 pourrait normaliser. Les chercheurs ont donc émis l'hypothèse que la perte ou l'inhibition du SGLT2 modifie les voies métaboliques au sein des cellules rénales afin de produire des métabolites qui protègent le rein. L'objectif de cette étude était de tester cette hypothèse à l'aide d'analyses métabolomiques, transcriptomiques et épigénétiques afin d'identifier les changements métaboliques dans les cellules tubulaires proximales (PTC) dépourvues de la fonction SGLT2 et de déterminer si ces voies contribuent à la protection rénale [17].
Méthodes. Des souris mâles de type sauvage (WT) et des souris présentant une perte de fonction du gène SGLT2 (« Sweet Pee » [SP]) ont été nourries soit avec un régime alimentaire normal, soit avec un régime riche en graisses (HFD) pendant une période pouvant aller jusqu’à 18 semaines, afin de provoquer un stress métabolique et une néphropathie diabétique précoce. La fonction rénale, l'histologie et les paramètres métaboliques ont été évalués en mesurant la créatinine sérique et le rapport albumine/créatinine urinaire, ainsi qu'en réalisant une immunocoloration du tissu rénal pour détecter des marqueurs de lésion et de fibrose. Un séquençage d'ARN unicellulaire (scRNA-seq) a été réalisé sur des cellules tubulaires proximales, et un profilage métabolomique non ciblé a été effectué sur le cortex rénal et le sérum. Un profilage épigénétique CUT&RUN a été réalisé pour mesurer les modifications de la méthylation des histones associées à l'altération des niveaux de métabolites.
Résultats. Chez les souris soumises à un régime riche en graisses, celles dépourvues de la fonction SGLT2 présentaient moins de lésions rénales, d’inflammation, de fibrose et de protéinurie que les souris de type sauvage, malgré un stress métabolique similaire. Le séquençage d’ARN unicellulaire a permis d’identifier une population de cellules tubulaires proximales lésées qui apparaissait dans les reins de type sauvage soumis à un régime riche en graisses, mais qui était largement supprimée en l’absence de la fonction SGLT2. L'analyse métabolomique a révélé que les reins dépourvus de SGLT2 présentaient une activité accrue du métabolisme de la méthionine, en particulier des taux plus élevés du métabolite S-adénosylméthionine (SAM) (Figure 31). Des expériences fonctionnelles ont montré que le blocage de l'enzyme MAT2A, responsable de la production de SAM, éliminait les effets protecteurs de la perte de SGLT2, tandis qu'une supplémentation en SAM réduisait les réponses inflammatoires dans des cellules rénales en culture exposées à un stress métabolique (Figure 32). L'inhibition pharmacologique de SGLT2 a produit des effets protecteurs similaires à ceux observés lors de la perte génétique de SGLT2. Enfin, des analyses épigénétiques ont montré qu'une élévation de la SAM augmentait la méthylation répressive des histones (H3K27me3) au niveau des promoteurs de gènes inflammatoires, y compris ceux de la voie NF-κB, ce qui entraînait une réduction de la signalisation inflammatoire. Ensemble, ces résultats indiquent que l'inhibition de SGLT2 favorise la protection rénale en augmentant la répression épigénétique des voies inflammatoires médiée par la SAM lors d'un stress métabolique.
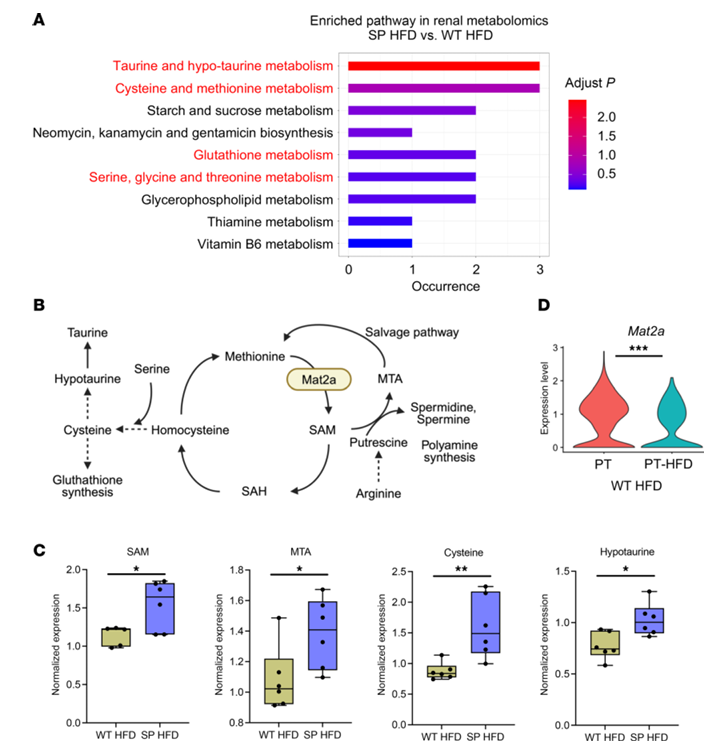
Figure 31. Profils métaboliques dans le rein. (A) Analyse des voies d'enrichissement métabolique dans des tissus isolés du cortex rénal. La couleur indique la valeur P ajustée. (B) Métabolisme de la méthionine et voies métaboliques. (C) Expression relative de SAM, MTA et des métabolites liés au métabolisme de la cystéine et de la taurine dans le cortex rénal (n = 6 par groupe). Les boîtes indiquent les 25e et 75e centiles, les lignes centrales indiquent les médianes, les moustaches s'étendent jusqu'aux valeurs minimales et maximales, et tous les points de données sont représentés. (D) Graphique en violon illustrant l'expression de Mat2a dans les cellules du tubule proximal (PT) normales et dans la sous-population de PTC (PT-HFD) apparue chez les souris de type sauvage nourries avec un régime riche en graisses (WT HFD). Image reproduite à partir de Maekawa et al., J Clin Invest, 2025, sous licence CC BY 4.0.
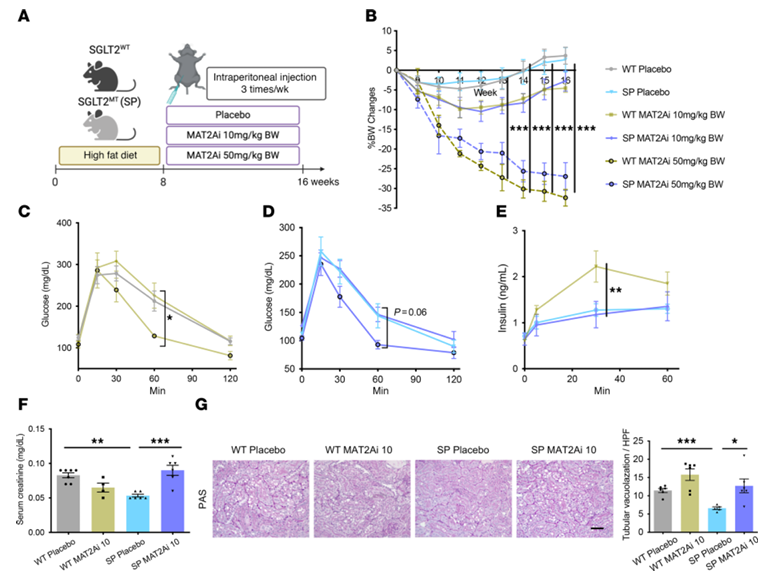
Figure 32. L'inhibition de l'enzyme MAT2A, codée par le gène de la méthionine, annule la protection rénale chez les souris SPHFD. (A) Schéma du protocole. (B) Évolution chronologique du pourcentage de variation du poids corporel. (C–E) Une faible dose d'inhibiteur de MAT2A (MAT2Ai) ne modifie pas la tolérance au glucose ni la capacité de sécrétion d'insuline, mais une dose élevée de MAT2Ai les réduit. (F) Taux de créatinine sérique. (G) Images représentatives de la coloration PAS. Image reproduite à partir de Maekawa et al., J Clin Invest, 2025, sous licence CC BY 4.0
Conclusions de l'étude
- Cette étude démontre que les métabolites jouent un rôle régulateur actif dans la progression de l'insuffisance rénale chronique et suggère que le ciblage du métabolisme de la méthionine ou des voies liées à la SAM pourrait constituer une stratégie thérapeutique viable pour les patients qui ne tolèrent pas les inhibiteurs du SGLT2.
- La métabolomique a joué un rôle crucial dans la caractérisation des liens entre l'inhibition des SGLT2, le métabolisme rénal et la réponse inflammatoire chez la souris, ce qui peut servir de point de départ à de futures études chez l'homme.
Identification d'un traitement potentiellement préventif pour une tumeur maligne agressive
Introduction. L'adénocarcinome canalaire pancréatique (PDAC) est la troisième cause de décès par cancer dans le monde et l'un des rares cancers dont la prévalence augmente à l'échelle mondiale. Actuellement, le taux de survie à cinq ans s'élève à 13 %, ce qui s'explique en grande partie par le fait que la plupart des diagnostics sont posés à un stade avancé, après que la tumeur a métastasé et que la résection n'est plus envisageable. Plus de 90 % des tumeurs PDAC contiennent des mutations activatrices du gène KRAS, ce qui fait de KRAS un moteur central de la croissance tumorale et une cible thérapeutique intéressante. Cependant, les réponses cliniques aux inhibiteurs de KRAS et aux médicaments ciblant la voie de signalisation KRAS (tels que les inhibiteurs RAF/MEK/ERK) ont été limitées, avec seulement des réponses partielles et un développement rapide d’une résistance aux médicaments. La plupart des stratégies de combinaison actuelles se concentrent sur des composants de la voie de signalisation KRAS canonique, mais les cellules cancéreuses peuvent contourner ces cibles grâce à des mécanismes compensatoires, ce qui souligne la nécessité d'identifier de nouvelles vulnérabilités en dehors de la voie KRAS traditionnelle.
Données préliminaires et objectifs de l'étude. Afin de répondre à ce besoin non satisfait, les chercheurs ont cherché à identifier les gènes essentiels à la survie des cellules du cancer du pancréas (PDAC) présentant une mutation du gène KRAS, dans le but de découvrir de nouvelles cibles thérapeutiques et des stratégies d'association susceptibles d'améliorer l'efficacité des inhibiteurs de KRAS dans le cancer du pancréas [18].
Méthodes. Des criblages de gènes inactivés par CRISPR/Cas9 à l'échelle du génome, des ensembles de données sur l'interférence ARN et de vastes bases de données sur la dépendance au cancer ont été utilisés pour identifier les gènes qui sont essentiels de manière sélective pour les cellules de PDAC porteuses d'une mutation KRAS. Des criblages de sensibilité aux médicaments ont été réalisés afin d'identifier les composés qui inhibent préférentiellement les cellules porteuses d'une mutation KRAS. Les gènes candidats ont été validés à l'aide d'approches de knockdown ou de knock-out génique et de tests de viabilité cellulaire dans des lignées cellulaires de PDAC. Le séquençage d'ARN, la métabolomique, la protéomique et le marquage par isotopes stables ont été utilisés pour analyser les modifications du métabolisme des nucléotides et des voies cellulaires après inhibition génique ou blocage de KRAS. L'importance fonctionnelle des cibles a été testée plus en détail à l'aide de modèles murins génétiquement modifiés et de modèles de transplantation tumorale orthotopique. Des composés PROTAC (Proteolysis-Targeting Chimera) dégradant sélectivement l'IMPDH2 ont été utilisés pour tester leur potentiel thérapeutique dans des organoïdes dérivés de patients et des modèles de tumeurs xénogreffées.
Résultats. La biosynthèse de novo des nucléotides guaniniques (DNGB) a été identifiée comme une vulnérabilité métabolique clé dans les cellules de PDAC présentant une mutation du gène KRAS, le gène critique étant IMPDH2, qui code pour une enzyme essentielle à la DNGB. La suppression pharmacologique de l'activité de l'IMPDH2 ou d'autres enzymes de la DNGB a considérablement réduit la croissance des cellules de PDAC. Dans des modèles murins, la délétion génétique de l'Impdh2 a réduit l'apparition de tumeurs pancréatiques, ralenti la progression de la maladie et amélioré la survie (Figure 33). Des analyses métabolomiques ont montré que le gène KRAS mutant stimule la biosynthèse des purines afin d'augmenter préférentiellement la production de nucléotides adéniques par rapport aux nucléotides guaniniques, créant ainsi un « goulot d'étranglement » métabolique pour la synthèse de la guanine (Figure 34). En conséquence, les cellules PDAC deviennent dépendantes de l'IMPDH2 pour maintenir leurs niveaux de nucléotides guaniniques et continuer à proliférer. L'inhibition de l'IMPDH2 a entraîné une déplétion des nucléotides guaniniques, ce qui a entravé la croissance des cellules PDAC. Il est important de noter qu'il a été démontré que l'expression de l'IMPDH2 n'était pas régulée par la voie de signalisation canonique KRAS, ce qui signifie que les cellules cancéreuses ne pouvaient pas compenser l'inhibition de l'IMPDH2 par la signalisation KRAS. Un composé dégradant sélectivement l'IMPDH2 (MED-B-4) a inhibé la croissance des lignées cellulaires PDAC avec une efficacité supérieure à celle des inhibiteurs de l'IMPDH existants.
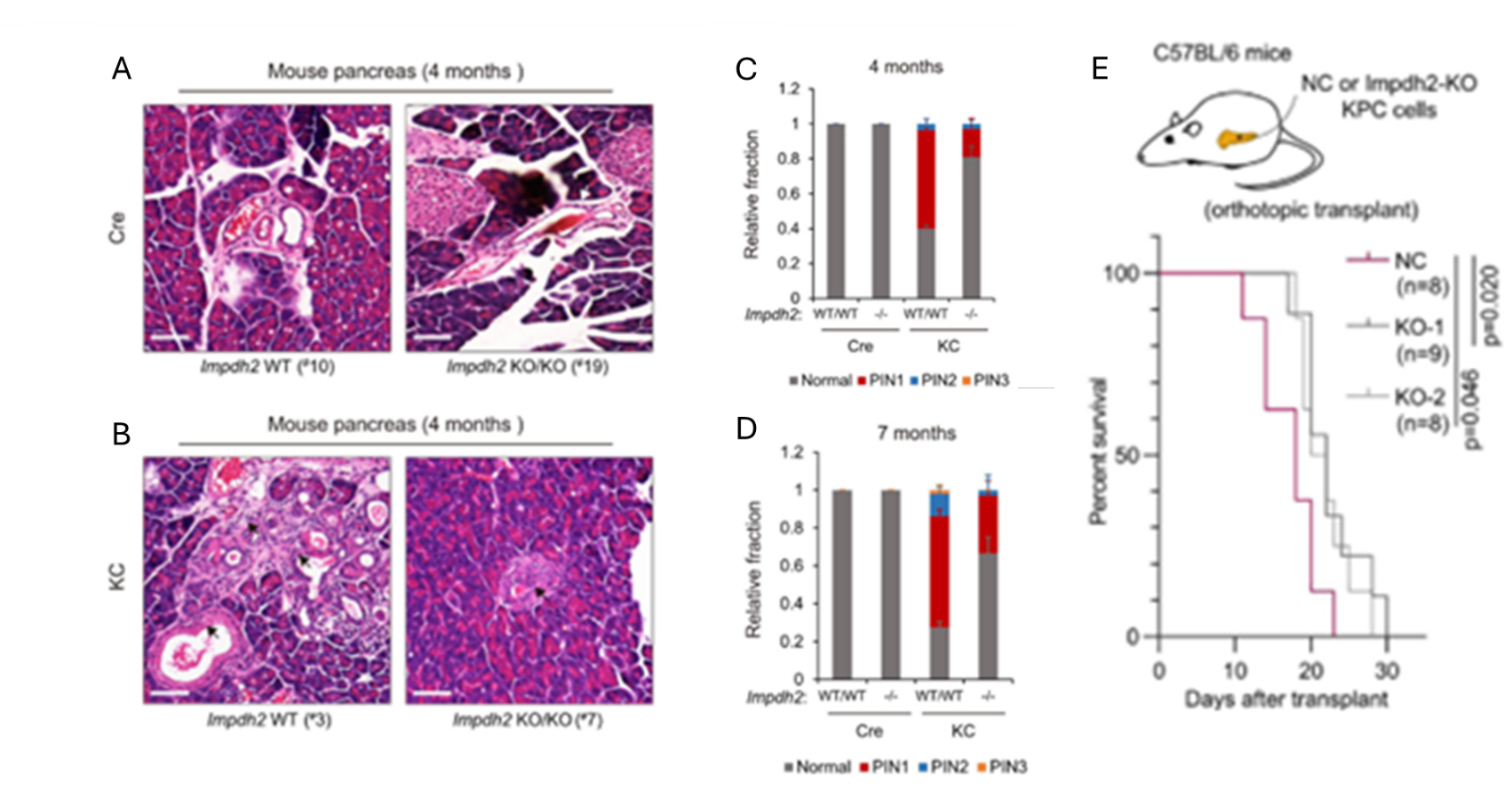
Figure 33. L'inhibition de la DNGB ou l'inactivation conditionnelle de l'Impdh2 inhibe la carcinogenèse pancréatique et la progression du cancer in vivo. Images représentatives de colorations H&E sur des échantillons de tissu pancréatique provenant de souris témoins et de souris (Cre; Impdh2-/-) (A), ou de souris KC et de souris (KC; Impdh2-/-) (B). Fréquence des stades de lésions de néoplasie intraépithéliale pancréatique (PanIN ou PIN) (PIN 1, PIN 2 et PIN 3) chez des souris de différents génotypes âgées de 4 mois (C) ou de 7 mois (D). Courbe de survie globale des souris ayant reçu une greffe orthotopique de cellules KPC témoins négatives, d'une lignée cellulaire PDAC transformée ou de cellules KPC knock-out pour Impdh2 (E). Image reproduite à partir de Wu et al., Gut, 2026, sous licence CC BY 4.0.
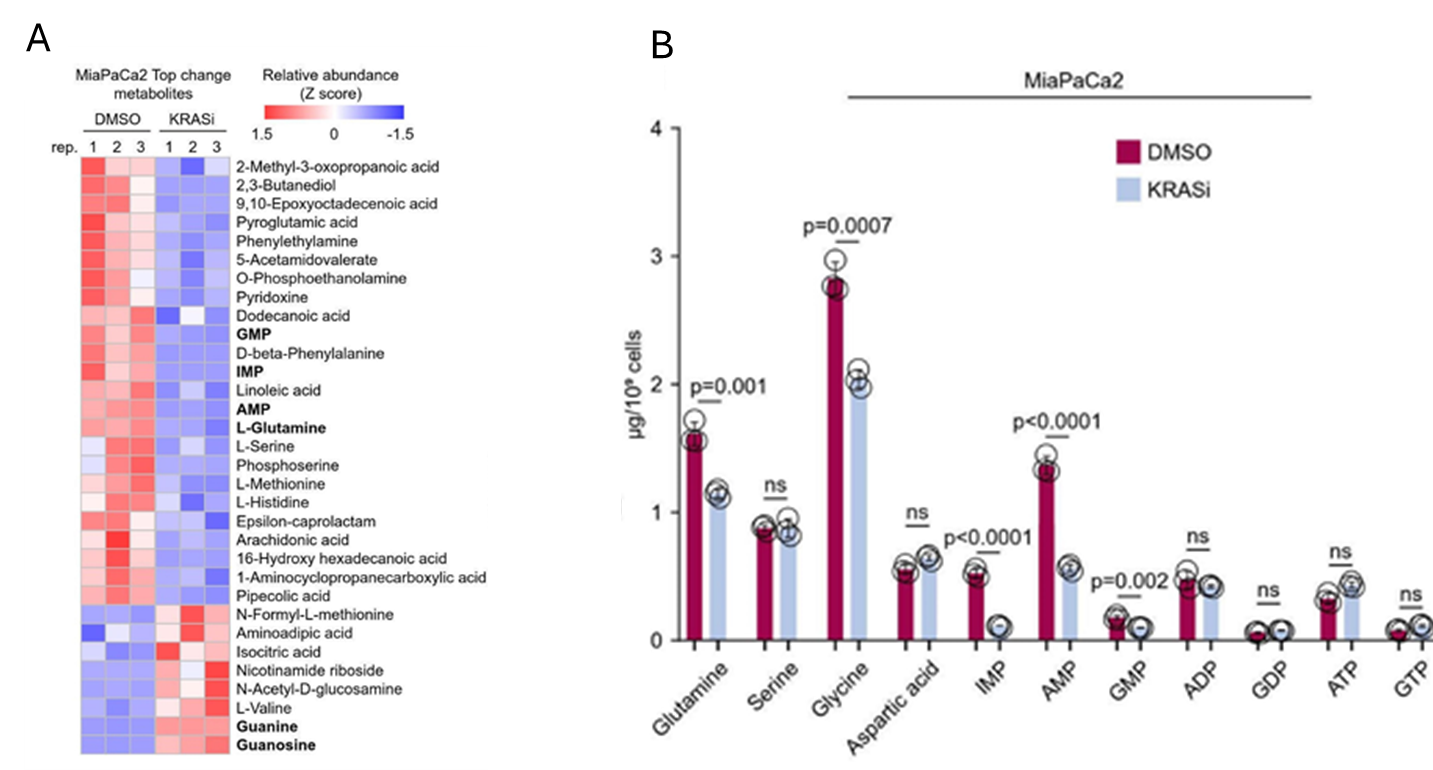
Figure 34. Le KRAS mutant stimule davantage la biosynthèse de novo des nucléotides adéniques que celle des nucléotides guaniniques. (A) Carte thermique illustrant les métabolites qui subissent des modifications significatives et constantes lors de l'inhibition aiguë du KRAS mutant (G12C) dans la lignée cellulaire PDAC transformée, les cellules MiaPaCa2, telle que déterminée par des analyses métabolomiques non ciblées. (B) Quantification des acides aminés et des nucléotides puriques par LC-MS/MS ciblée dans les cellules témoins et les cellules soumises à une inhibition de KRAS (témoin : DMSO pendant 120 min ; inhibition de KRAS : 100 nM d'AMG510 pendant 120 min). Image reproduite à partir de Wu et al., Gut, 2026, sous licence CC BY 4.0.
Conclusions de l'étude
- Avant cette étude, la biosynthèse de novo des nucléotides guaniniques était une vulnérabilité métabolique sous-estimée dans le cancer du pancréas (PDAC) présentant une mutation du gène KRAS. La métabolomique a joué un rôle important en démontrant que la mutation KRAS augmente de manière spécifique la production de nucléotides adéniniques, ce qui entraîne une pénurie de nucléotides guaniniques et rend les cellules du PDAC dépendantes de l'IMPDH2, l'enzyme clé chargée de convertir l'inosine monophosphate en nucléotides guaniniques.
- Il est important de noter que, comme l'IMPDH2 n'est pas régulée par la voie de signalisation canonique KRAS, les cellules du cancer du pancréas (PDAC) ne peuvent pas compenser son inhibition par les mécanismes de rétroaction habituels de la voie KRAS. Cela a permis de découvrir que le blocage de l'IMPDH2 entraîne une déplétion irréversible en nucléotides guaniniques, ce qui perturbe la synthèse de l'ADN et de l'ARN et freine la croissance tumorale.
- Ces résultats ont des implications pour les traitements futurs, car ils suggèrent que la dégradation sélective de l'IMPDH, plutôt qu'une inhibition généralisée, pourrait constituer une stratégie plus ciblée pour le traitement du cancer du pancréas.
Microbiome
Mise en lumière des mécanismes reliant le microbiome à la sclérose en plaques
Introduction. Il a été démontré que les métabolites microbiens influencent la régulation immunitaire, l'inflammation et les fonctions neurologiques, ce qui laisse supposer qu'ils pourraient contribuer à l'activité ou à la progression de la maladie. Diverses études ont mis en évidence des anomalies dans le microbiome intestinal des patients atteints de sclérose en plaques (SEP), mais le rôle des microbes intestinaux et de leurs métabolites dans la pathologie de la SEP reste à établir.
Données préliminaires et objectifs de l'étude. L'objectif de cette étude était de déterminer si certains microbes intestinaux et certains métabolites fécaux circulants étaient associés à une aggravation de la sclérose en plaques (SEP) ou à l'évolution vers une forme progressive de la maladie, en analysant les données cliniques, microbiologiques et métabolomiques longitudinales provenant de participants à une cohorte de patients atteints de SEP bien caractérisée [19].
Méthodes. Cette étude a porté sur les participants de la cohorte CLIMB (Comprehensive Longitudinal Investigation of Multiple Sclerosis) et a analysé les données cliniques associées, les images IRM ainsi que les échantillons de plasma et de selles conservés en banque. Les chercheurs ont suivi les patients pendant environ deux ans et les ont classés en trois groupes définis par : 1) une maladie stable, 2) une aggravation du handicap, et 3) une transition de la SEP récurrente-rémittente vers la SEP progressive. Un séquençage métagénomique de type « shotgun » a été réalisé sur des échantillons de selles afin de caractériser le microbiote intestinal. Les échantillons de selles et de sérum ont été analysés à l'aide d'un profilage métabolomique global afin de mesurer les métabolites d'origine microbienne et ceux provenant de l'hôte. Des modèles statistiques ont été utilisés pour identifier les associations entre les taxons microbiens, les niveaux de métabolites et les changements dans les résultats cliniques, notamment la progression du handicap, les résultats d'IRM et les mesures de la qualité de vie.
Résultats. Des associations significatives ont été mises en évidence entre la composition du microbiote intestinal, les profils métaboliques et la progression de la SEP. Les personnes ayant connu une aggravation de la maladie ou une évolution vers une SEP progressive présentaient des niveaux réduits de plusieurs taxons microbiens bénéfiques, notamment des espèces connues pour produire des acides gras à chaîne courte et d’autres métabolites anti-inflammatoires (figure 35). À l'inverse, des niveaux plus élevés de métabolites produits par ces microbes bénéfiques étaient associés à une maladie stable. Les caractéristiques microbiennes et les niveaux de métabolites étaient associés au score d'invalidité, aux indicateurs IRM de l'activité de la maladie et aux résultats rapportés par les patients, ce qui indique que les changements métaboliques dérivés du microbiome pourraient jouer un rôle direct dans la progression de la SEP (Figure 36).
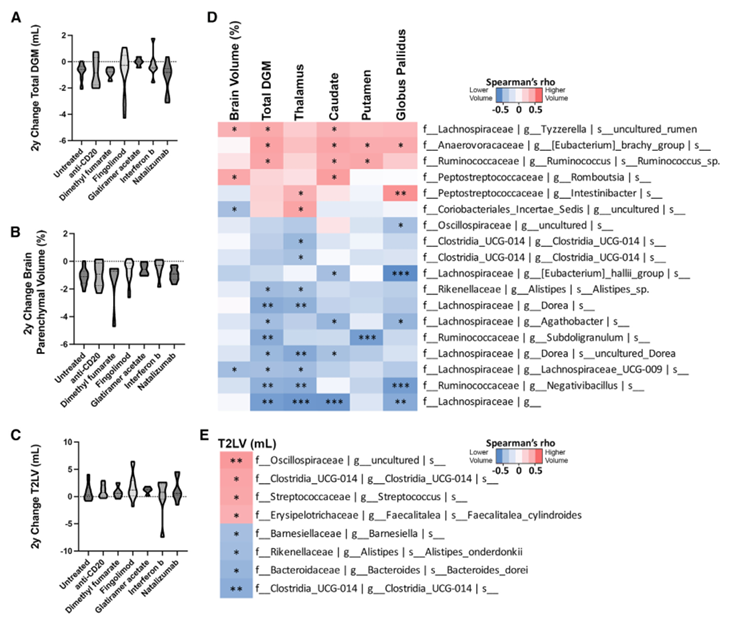
Figure 35. Les microbes intestinaux sont en corrélation avec les variations sur deux ans observées en IRM 3T. Évolution du DGM (A), du volume parenchymateux total du cerveau (B) et du T2LV stratifiée en fonction du DMT (C). Les corrélations de Spearman montrent que certains taxons microbiens présentent une corrélation significative positive (rouge) ou négative (bleu) avec les variations sur deux ans des mesures volumétriques en IRM 3T (D). La corrélation de Spearman montre que les taxons sont positivement (rouge) ou négativement (bleu) corrélés au T2LV (E). Image reproduite à partir de Schwerdtfeger et al., Cell Rep Med, 2025, sous licence CC BY 4.0.
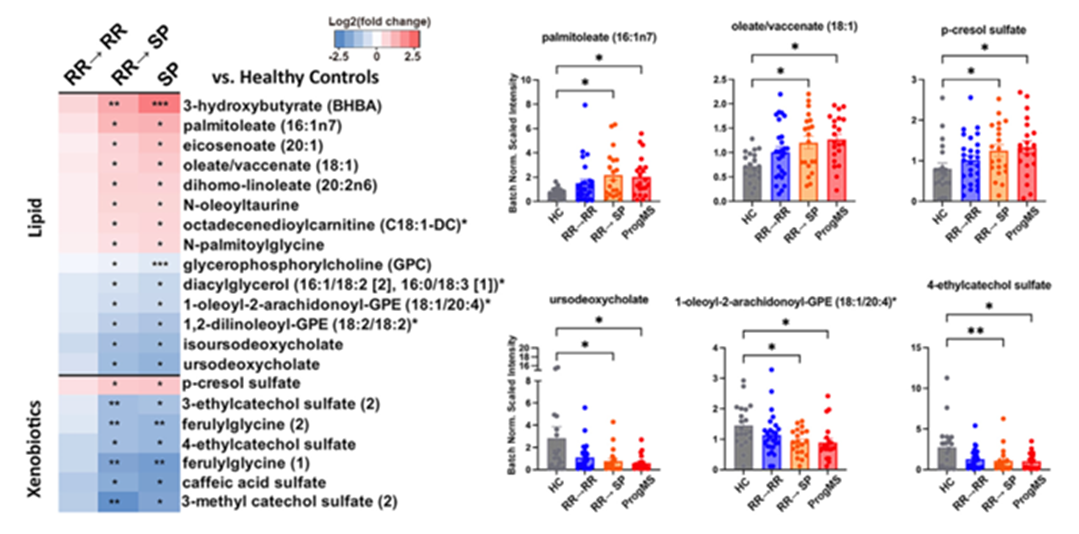
Figure 36. Métabolites sériques associés au développement d'une SEP progressive. (A) Ensemble des métabolites sériques présentant des variations significatives chez les patients ayant évolué d'une forme récurrente-rémittente (RR) vers une forme progressive stable (SP). (B) Diagrammes à barres présentant les valeurs individuelles des altérations métaboliques biologiquement significatives en fonction du stade de la maladie. Image reproduite à partir de Schwerdtfeger et al., Cell Rep Med, 2025, sous licence CC BY 4.0
Conclusions de l'étude
- Les résultats de cette étude viennent étayer l'hypothèse selon laquelle le microbiome intestinal pourrait influencer la sclérose en plaques par le biais de voies de signalisation métaboliques et de la modulation immunitaire ; les taxons microbiens et les métabolites identifiés pourraient servir de biomarqueurs de la progression de la maladie ou constituer de nouvelles cibles thérapeutiques susceptibles d'être modulées par l'alimentation.
- Sans la métabolomique, les chercheurs auraient seulement pu observer des changements dans la composition des espèces microbiennes et n'auraient pas pu déterminer quelles voies métaboliques étaient perturbées, quelles molécules bioactives pouvaient influencer la signalisation immunitaire et la neuroinflammation, ni quelles molécules méritaient d'être étudiées plus en détail en tant que cibles thérapeutiques potentielles.
Comprendre l'influence du microbiome sur la physiopathologie du diabète de type 2
Introduction. Le diabète de type 2 (DT2) est une maladie métabolique complexe influencée par des facteurs génétiques, environnementaux et microbiens. De plus en plus de données suggèrent que le microbiome intestinal contribue à la santé métabolique en produisant des métabolites bioactifs qui agissent sur la physiologie de l'hôte, notamment le métabolisme du glucose et l'inflammation. Bien que des études de séquençage du microbiome aient permis d'identifier des taxons microbiens associés au DT2, elles n'expliquent pas entièrement les mécanismes biochimiques fonctionnels reliant les microbes intestinaux aux maladies métaboliques.
Données préliminaires et objectifs de l'étude. Étant donné que les métabolites microbiens agissent comme des molécules de signalisation qui influencent le métabolisme de l'hôte, les chercheurs ont cherché à déterminer comment l'activité métabolique microbienne contribue au risque et à la progression du DT2 en recourant au séquençage métagénomique et à des analyses métabolomiques globales [20].
Méthodes. Cette étude a analysé des cohortes dont le statut diabétique et les données de phénotypage métabolique étaient connus. Le séquençage métagénomique a été utilisé pour caractériser la composition et l'abondance du microbiome intestinal. Le plasma et les selles ont été analysés par profilage métabolomique global afin de mesurer un large éventail de métabolites dérivés de l'hôte et des microbes. Des analyses statistiques ont été utilisées pour identifier les métabolites et les taxons microbiens associés au DT2 et aux traits métaboliques connexes, notamment la résistance à l'insuline et le contrôle glycémique. Des analyses intégratives ont été utilisées pour identifier les liens entre des microbes et des métabolites spécifiques afin de déterminer comment les voies métaboliques microbiennes influencent les phénotypes métaboliques de l'hôte.
Résultats. Les personnes atteintes de DT2 présentaient des profils microbiologiques et métaboliques distincts de ceux des personnes en bonne santé. Plusieurs taxons microbiens ont été associés à des concentrations modifiées de métabolites impliqués dans le métabolisme des acides aminés, le métabolisme des lipides et les voies de fermentation microbienne. La métabolomique a mis en évidence des métabolites d'origine microbienne spécifiques qui présentaient une corrélation significative avec les mesures de l'insulinorésistance et de la régulation du glucose (Figure 37). Il est intéressant de noter que les métabolites associés au DT2 se sont inversés en réponse à une intervention diététique ou à la pratique d'une activité physique (Figure 38). Certains métabolites ont montré des associations plus fortes avec le risque de DT2 que les taxons microbiens eux-mêmes, ce qui suggère que l'activité métabolique microbienne, plutôt que la simple abondance microbienne, pourrait être un facteur clé des changements métaboliques liés à la maladie.
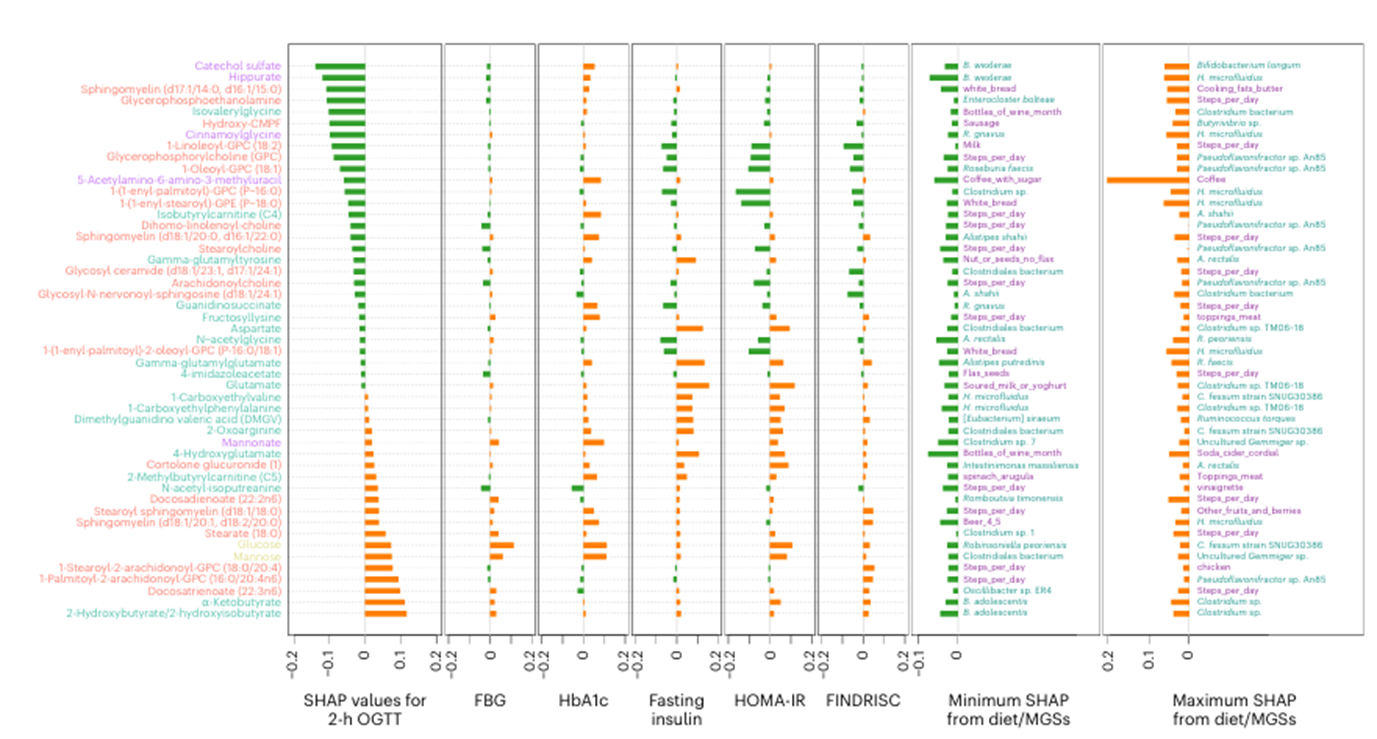
Figure 37. Caractéristiques de la flore intestinale expliquant l'intolérance au glucose. Les dix principaux métabolites identifiés comme des caractéristiques importantes dans le test de tolérance au glucose par voie orale (OGTT) de 2 heures, la glycémie à jeun (FBG), l'HbA1c, l'insuline à jeun, l'indice HOMA-IR (Homeostatic Model Assessment of Insulin Resistance) ou le score FINDRISC (Finnish Diabetes Risk Score) (n = 49). Image reproduite à partir de Wu et al., Nat Med, 2025, sous licence CC BY 4.0.
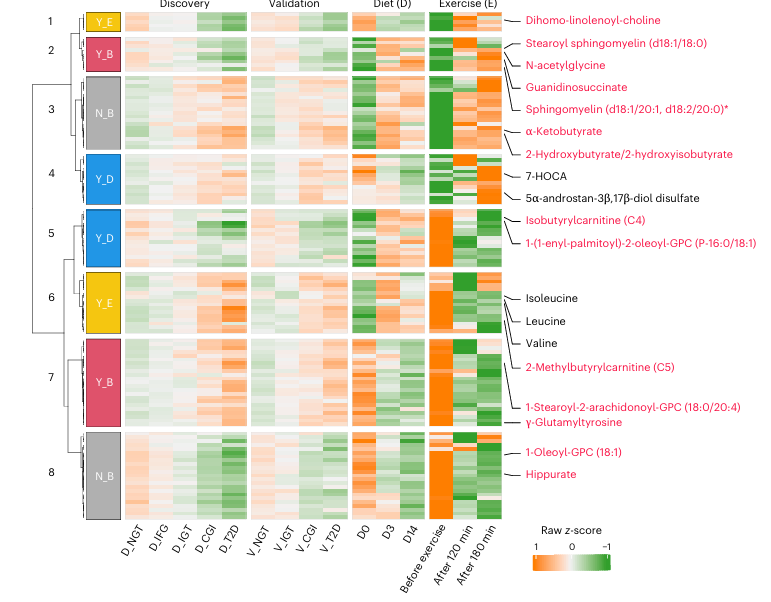
Figure 38. Réponses des métabolites associés au prédiabète et au DT2 à une intervention diététique de deux semaines ou avant et après l'exercice. Carte thermique montrant les métabolites communs impliqués dans le métabolisme des acides aminés, des lipides et des xénobiotiques (n = 123) dans deux essais cliniques portant soit sur un régime alimentaire (14 jours), soit sur une séance d'exercice d'une heure (avant, 120 et 180 minutes après l'exercice), par rapport aux 502 métabolites altérés observés dans le prédiabète et le DT2. Les réponses inversées (Y, oui ; N, non) par le régime alimentaire (D), l'exercice physique (E) ou les deux (B) ont été regroupées et sont représentées par des couleurs distinctes à côté des branches de regroupement des lignes. Les métabolites représentatifs sont indiqués en rouge, et cinq autres en noir. Image reproduite à partir de Wu et al., Nat Med, 2025, sous licence CC BY 4.0.
Un lien peu étudié entre les symptômes du syndrome du côlon irritable et les fonctions cognitives
Introduction. Le syndrome du côlon irritable (SCI) est un trouble gastro-intestinal caractérisé par des douleurs abdominales récurrentes et une altération des habitudes intestinales. Les interactions entre le cerveau, l'intestin et le microbiome sont de plus en plus reconnues comme des régulateurs importants de la fonction gastro-intestinale, de la perception des symptômes et de l'humeur, ce qui en fait des cibles privilégiées pour une intervention thérapeutique dans le cas du SCI. La thérapie cognitivo-comportementale (TCC) est une intervention efficace ciblant le cerveau qui enseigne des compétences de traitement de l'information afin de traiter les facteurs psychologiques connus pour exacerber les symptômes abdominaux, notamment les stratégies d'adaptation inadaptées, l'inquiétude intense et la réactivité au stress.
Données préliminaires et objectifs de l'étude. Un groupe de recherche avait précédemment démontré, dans le cadre d'un vaste essai clinique randomisé, que deux programmes de TCC adaptés au syndrome du côlon irritable (SCI) permettaient d'obtenir une amélioration durable des symptômes gastro-intestinaux, par rapport à un programme d'éducation sur le SCI qui servait de groupe témoin pour contrôler les effets non spécifiques liés au fait de suivre un traitement. Sur la base de ces résultats, l'équipe de recherche a émis l'hypothèse que la TCC atténue les symptômes en modulant principalement la composante cérébrale de l'axe cerveau-intestin-microbiome (CIM) et que les signaux microbiens envoyés au cerveau sous forme de métabolites neuroactifs, notamment les acides gras à chaîne courte et la sérotonine, pourraient moduler la réactivité aux effets biologiques de la TCC. Ainsi, l'objectif de cette étude était de déterminer si les signatures métaboliques chez les patients atteints du SCI sont associées aux performances cognitives et aux symptômes liés à l'axe C-I-M [21].
Méthodes. Les patients éligibles ont été randomisés pour recevoir soit 10 séances de TCC en cabinet, soit 4 séances de TCC principalement à domicile avec un contact minimal avec le thérapeute, au cours d’une phase aiguë de 10 semaines. Tous les participants ont subi des examens IRM de référence et post-traitement. Des questionnaires cliniques ont été utilisés pour évaluer la gravité des symptômes gastro-intestinaux. Les échantillons de selles ont été analysés par séquençage du gène 16S rRNA et par métabolomique non ciblée. Les analyses statistiques ont permis d'identifier les métabolites associés au statut du SCI, aux performances cognitives et à la gravité des symptômes.
Résultats. Sur les 84 participants atteints du syndrome du côlon irritable (SCI) ayant subi une imagerie cérébrale, 58 ont été classés comme répondeurs à la TCC, tandis que 26 ont été classés comme non-répondeurs, sur la base d’une diminution d’au moins 50 points sur l’échelle d’évaluation de la gravité des symptômes du SCI après le traitement. La diversité bêta microbienne présentait une différence significative entre les répondeurs et les non-répondeurs, et l’abondance de la sérotonine, un neurotransmetteur d’origine microbienne, était significativement plus élevée chez les répondeurs. À la suite de la TCC, les répondeurs ont présenté une diminution de la connectivité entre plusieurs régions associées à des réseaux cérébraux spécifiques liés à la régulation émotionnelle (Figure 39). Il est à noter que ces changements s’accompagnaient d’une diminution significative tant des douleurs abdominales que du stress perçu. Parmi les patients ayant répondu à la TCC, les Bacteroides étaient plus abondants au départ par rapport aux non-répondeurs (Figure 40). Un classificateur de type « random forest » comprenant 11 genres bactériens a été utilisé pour prédire la réponse à la TCC. Ce classificateur a permis de prédire avec précision la réponse à la TCC, démontrant ainsi que le microbiome peut servir de biomarqueur efficace pour évaluer la réponse potentielle d'un patient au traitement et pour identifier en priorité les patients susceptibles de bien répondre à la TCC.
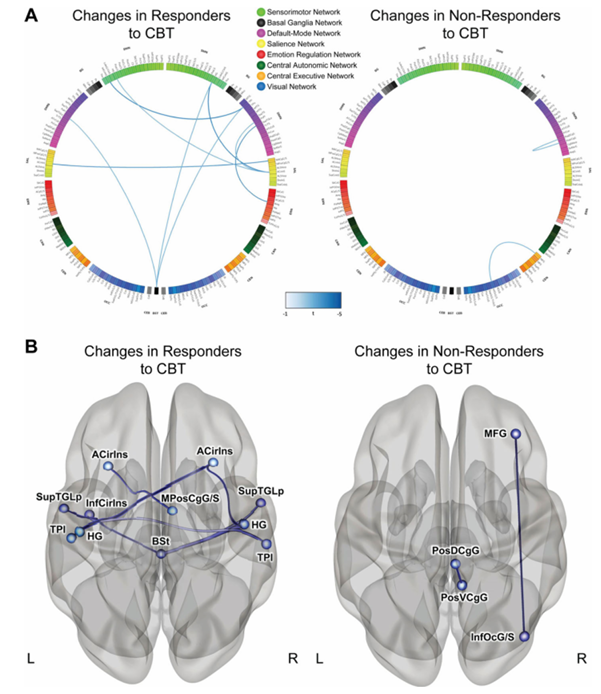
Figure 39. Évolution de la connectivité fonctionnelle chez les patients répondant et ne répondant pas à la TCC. (A) Connectogrammes illustrant les différences de connectivité par paires entre les groupes. Les lignes bleues indiquent des baisses significatives de la connectivité. (B) Régions présentant des différences significatives entre les patients répondant et ne répondant pas à la TCC. Image reproduite à partir de Jacobs et al., Microbiome, 2021, sous licence CC BY 4.0.
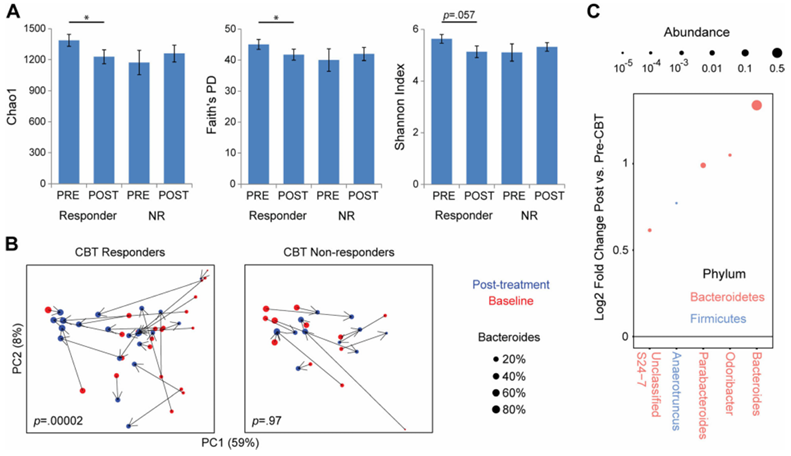
Figure 40. Les patients ayant répondu à la TCC présentent une modification de la composition du microbiome intestinal après la TCC, caractérisée par une expansion des Bacteroides. (A) La diversité alpha microbienne fécale est représentée pour les patients ayant répondu à la TCC et ceux n'y ayant pas répondu (NR) au départ (PRE) et après la TCC (POST). (B) Analyse en coordonnées principales des données de séquences d'ARNr 16S avant et après la TCC, stratifiées selon le statut de réponse à la TCC. Chaque point représente un échantillon, coloré en fonction du moment (rouge = début de l'étude, bleu = après la TCC) et dont la taille correspond à l'abondance de Bacteroides. (C) Les genres microbiens présentant une association statistiquement significative avec le statut de répondeur à la TCC sont indiqués. Image reproduite à partir de Jacobs et al., Microbiome, 2021, sous licence CC BY 4.0.
Conclusions de l'étude
- Ces résultats suggèrent l'existence d'un mécanisme lié au microbiome par lequel les patients atteints du syndrome du côlon irritable ressentent des douleurs en l'absence de pathologie, et ouvrent de nouvelles perspectives quant à l'utilisation de certains composants de l'axe BGM comme biomarqueurs pour le traitement de ce syndrome.
- La métabolomique a joué un rôle essentiel dans cette étude, car elle a permis de mesurer directement les molécules de signalisation entre l'intestin et le cerveau qui relient les troubles gastro-intestinaux aux symptômes cognitifs dans le syndrome du côlon irritable, contribuant ainsi à élucider le lien complexe entre le microbiome et la santé humaine.
Santé de la population
Identifier de nouveaux liens entre les voies biochimiques et les maladies non transmissibles afin d'expliquer la multimorbidité
Introduction. La multimorbidité, c'est-à-dire la cooccurrence de plusieurs maladies chroniques, représente un fardeau sanitaire mondial considérable dont la prévalence ne cesse d'augmenter. Avant de pouvoir traiter ce problème de manière adéquate, nous devons mieux comprendre les facteurs de risque et les mécanismes de progression des maladies. Bien que de nombreuses affections chroniques partagent des facteurs de risque et des mécanismes biologiques communs, la majorité des études se limitent à une seule maladie. Le profilage moléculaire pourrait permettre d'identifier des voies biologiques communes qui prédisposent les individus à plusieurs maladies. À cette fin, les chercheurs ont réalisé un profilage métabolomique non ciblé sur des échantillons de plasma prélevés auprès de la cohorte Norfolk de l'étude EPIC (European Prospective Investigation into Cancer) [22].
Données préliminaires et objectifs de l'étude. L'objectif de cette étude était d'identifier les voies métaboliques communes, de déterminer quelles associations sont liées à des facteurs de risque modifiables et de mettre en évidence les métabolites associés au développement de plusieurs maladies chroniques.
Méthodes. Les échantillons de plasma provenant de 11 966 participants de la cohorte EPIC-Norfolk ont fait l'objet d'une analyse métabolomique non ciblée. Les maladies incidentes et la mortalité ont été identifiées grâce à un suivi à long terme utilisant des dossiers médicaux électroniques reliés, des données d'hospitalisation et des registres du cancer couvrant plus de 219 000 années-personnes. Les associations entre les taux de métabolites et l'incidence des maladies ont été évaluées à l'aide de modèles de risques proportionnels de Cox ajustés en fonction de l'âge et du sexe. Une régression linéaire a été utilisée pour examiner les associations entre les métabolites et la multimorbidité.
Résultats. Les résultats ont mis en évidence 458 métabolites associés à au moins une issue pathologique, ce qui a donné lieu à 1 226 associations métabolite-maladie au total. 65,5 % des métabolites significatifs étaient associés à au moins deux maladies, ce qui témoigne d’un chevauchement biologique important entre les affections chroniques. Des liens étroits ont été observés entre les maladies cardiométaboliques, respiratoires, rénales et hépatiques, et de nombreux métabolites ont présenté des directions d’association cohérentes dans plusieurs pathologies (figure 41). Les analyses de médiation ont montré que de nombreuses relations métabolite-maladie étaient associées à des facteurs de risque communs, notamment l'obésité, le tabagisme, l'inflammation, la résistance au glucose et des taux élevés de lipides. Certains métabolites étaient associés de manière unique à des maladies spécifiques, tandis que 30 métabolites étaient significativement liés au développement de la multimorbidité (Figure 42).
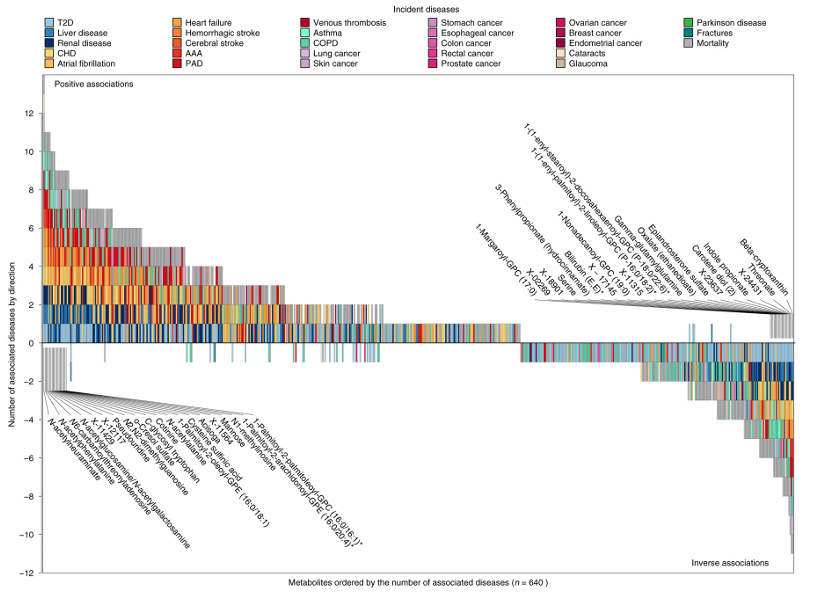
Figure 41. Diagramme en briques illustrant le classement des métabolites en fonction du nombre de critères de jugement cliniques associés. Une valeur de P < 0,001 a été considérée comme significative, en tenant compte des 28 maladies testées pour chaque métabolite. L'axe des x indique le rang de chaque métabolite en fonction du nombre de critères de jugement cliniques associés, les associations inverses étant comptabilisées comme des nombres négatifs afin de simplifier la représentation des résultats. L'axe des y indique le nombre de métabolites associés, les nombres positifs indiquant des associations positives et les nombres négatifs des associations inverses. Les couleurs de chaque case indiquent le critère de jugement de la maladie associé. Les métabolites sélectionnés présentant plusieurs critères de jugement associés sont annotés. Image reproduite à partir de Pietzner et al., Nat Med, 2021, sous licence CC BY 4.0.
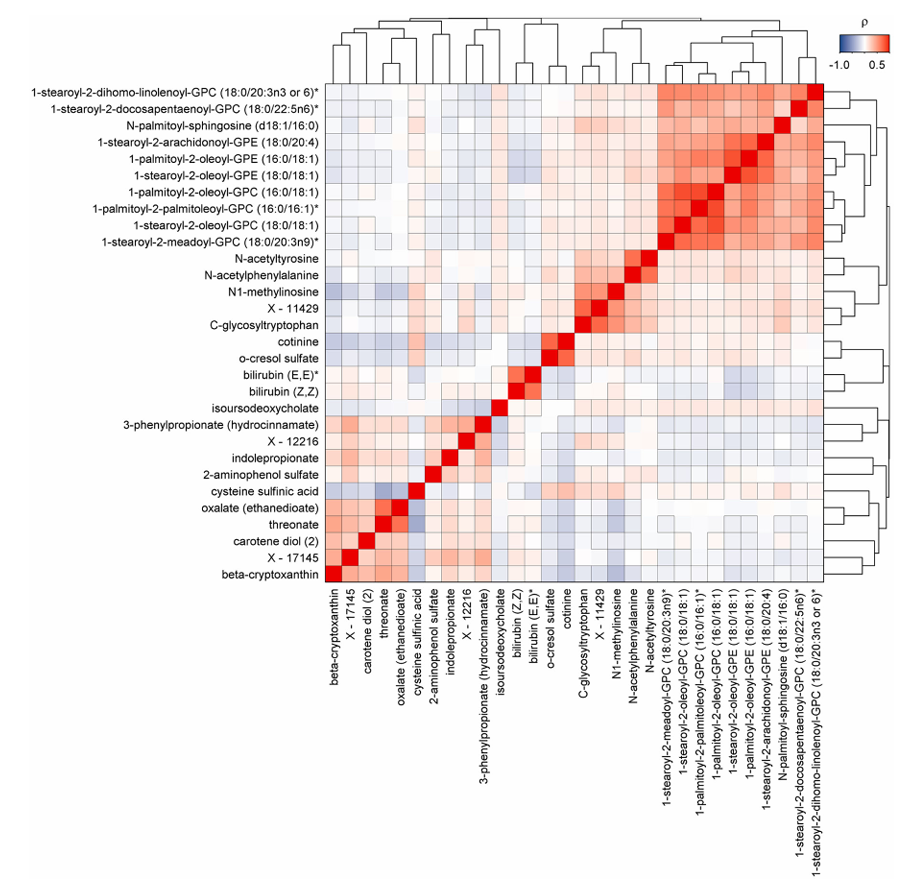
Figure 42. Carte thermique des corrélations par paires entre les métabolites candidats à la multimorbidité. Matrice de corrélation par paires des métabolites plasmatiques associés de manière significative à l'incidence de la multimorbidité liée aux maladies non transmissibles. Les couleurs indiquent des corrélations positives (rouge) ou inverses (bleu), et les cadres noirs indiquent une signification statistique après correction pour les tests multiples. Image reproduite à partir de Pietzner et al., Nat Med, 2021, sous licence CC BY 4.0.
Conclusions de l'étude
- Dans cette étude, la métabolomique non ciblée a joué un rôle déterminant dans l'identification des liens entre les voies biochimiques communes à plusieurs maladies, ce qui a permis à la fois de mieux comprendre les mécanismes sous-jacents au risque de maladie et de fournir des pistes concrètes pour le développement de futurs traitements.
- Dans de nombreux cas, les voies associées à plusieurs maladies présentaient les mêmes tendances d'association pour chacune de ces maladies prises individuellement. Cela laisse penser que les interventions futures pourraient cibler ces voies communes afin de prévenir plusieurs maladies.
- Les résultats de cette étude pourraient également aider à identifier des sous-types de multimorbidité en examinant comment ces mécanismes sont liés à la cooccurrence de maladies apparemment sans rapport entre elles.
Prédire l'âge biologique pour mieux évaluer les risques de maladie
Introduction. L'âge chronologique est un facteur de risque majeur pour de nombreuses maladies, mais il ne rend pas compte du processus complexe du vieillissement biologique ni de la grande variabilité de ce vieillissement d'un individu à l'autre. La différence entre le vieillissement chronologique et le vieillissement biologique pourrait constituer un indicateur plus pertinent de l'état de santé que le vieillissement chronologique seul. Le vieillissement biologique reflète des interactions complexes entre des facteurs génétiques, liés au mode de vie et environnementaux, et l'identification de biomarqueurs qui représentent mieux ce processus pourrait améliorer la compréhension des risques de maladie et des trajectoires de santé. La métabolomique est une approche prometteuse, car les métabolites circulants reflètent à la fois l'activité métabolique endogène et les expositions externes, notamment l'alimentation, les médicaments et les facteurs environnementaux.
Données préliminaires et objectifs de l'étude. Les études antérieures sur la prédiction de l'âge métabolique ont été limitées par la petite taille des échantillons, le caractère ciblé des panels de métabolites ou des tranches d'âge restreintes, ce qui peut réduire la précision prédictive et la généralisation des résultats. Par conséquent, les objectifs de cette étude étaient de développer un modèle métabolomique robuste de prédiction de l'âge en réalisant un profilage métabolomique non ciblé sur INTERVAL, une vaste cohorte basée sur la population, et d'évaluer si les estimations de l'âge métabolique ainsi obtenues étaient associées à des résultats de santé [23].
Méthodes. L'étude INTERVAL est une étude de cohorte prospective portant sur environ 50 000 participants, intégrée à un échantillon randomisé de donneurs de sang. Dans le cadre de cette étude, des échantillons de plasma provenant de 12 000 donneurs de sang en bonne santé, âgés de 18 à 75 ans, ont été analysés à l'aide de la métabolomique globale. La régression de Ridge combinée à la méthode du bootstrapping a été utilisée pour construire et valider en interne des modèles permettant de prédire l'âge chronologique à partir des profils métaboliques. Des modèles distincts ont également été construits en utilisant uniquement des métabolites endogènes et en exploitant des données spécifiques au sexe pour les hommes et les femmes. Les prédictions d'âge métabolomique ainsi obtenues ont ensuite été testées dans le cadre de l'étude néerlandaise sur l'épidémiologie de l'obésité (NEO) afin de déterminer si les différences entre l'âge métabolomique prédit et l'âge chronologique étaient associées à des caractéristiques de santé.
Résultats. Les modèles métabolomiques de prédiction de l'âge ont démontré une grande efficacité, expliquant plus de 80 % de la variation de l'âge chronologique lorsque les métabolites endogènes et xénobiotiques étaient pris en compte (figure 43). Parmi les métabolites clés ayant fortement contribué à la prédiction de l'âge figuraient l'hydroxyasparagine, le vanillylmandélate et la 5,6-dihydrouridine. Lorsque le modèle a été appliqué à la cohorte NEO, un âge métabolomique supérieur à l'âge chronologique a été associé à l'obésité et aux maladies cardiovasculaires, en partie en raison des métabolites xénobiotiques reflétant la prise de médicaments et des facteurs environnementaux tels que la cotinine (figure 44). L'étude a également identifié 163 métabolites présentant des différences entre les hommes et les femmes, et les modèles spécifiques au sexe ont montré une performance prédictive élevée mais une corrélation modérée entre eux, ce qui suggère que ces modèles ont permis de saisir des différences significatives entre les sexes dans les profils métaboliques liés à l'âge.
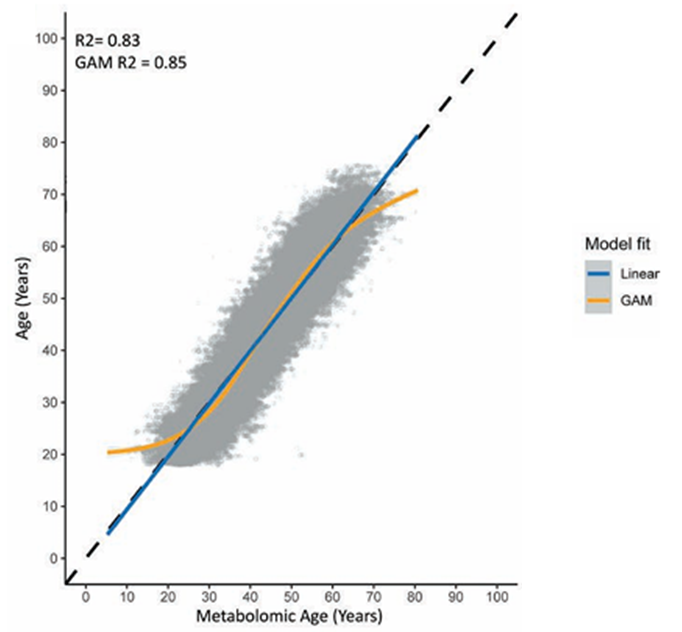
Figure 43. Graphique de corrélation représentant l'âge métabolomique (âge prédit) sur l'axe des x et l'âge chronologique sur l'axe des y pour l'ensemble des métabolites endogènes et xénobiotiques mesurés. Image reproduite à partir de Faquih et al., J Gerontol A Biol Sci Med Sci, 2025, sous licence CC BY 4.0.
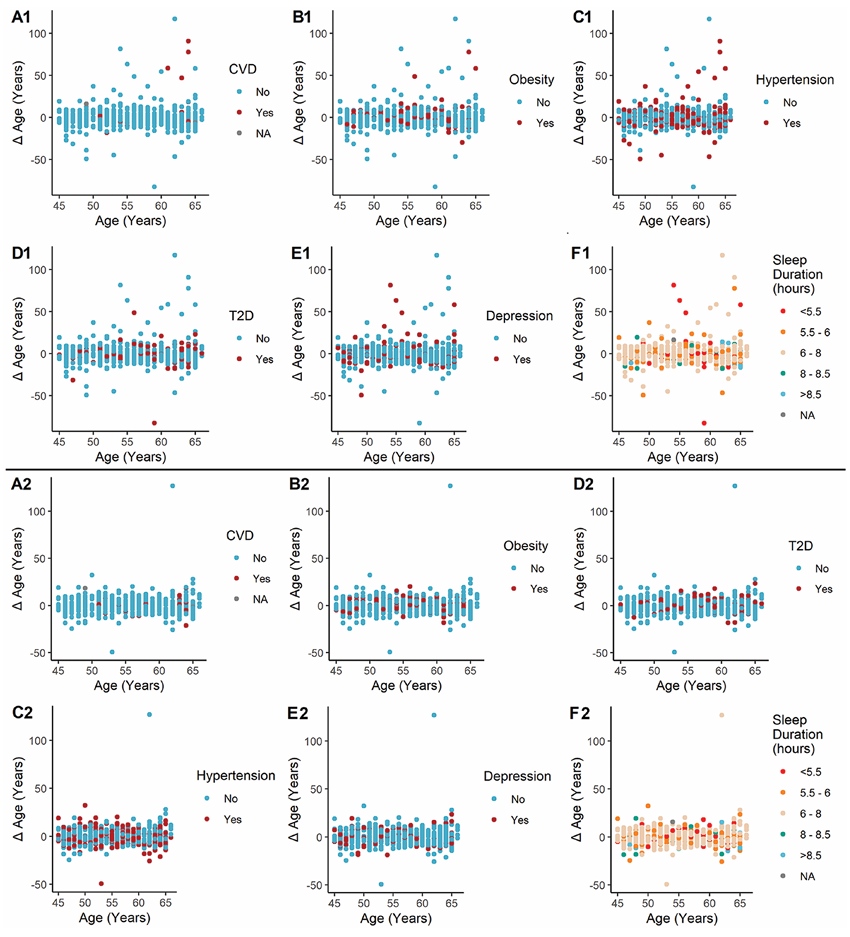
Figure 44. Diagrammes de dispersion illustrant la différence d'âge (Δ âge) telle qu'elle est prédite à l'aide du modèle combinant les métabolites endogènes et xénobiotiques (A1-F1) et du modèle basé uniquement sur les métabolites endogènes (A2-F2). NEO = Étude épidémiologique néerlandaise sur l'obésité. Image reproduite à partir de Faquih et al., J Gerontol A Biol Sci Med Sci, 2025, sous licence CC BY 4.0.
Conclusions de l'étude
- L'intégration d'un large éventail de métabolites endogènes et xénobiotiques, mesurés à l'aide de la métabolomique globale, a permis d'élaborer un modèle métabolomique d'âge robuste qui a surpassé de nombreuses approches antérieures.
- Ces résultats montrent que les modèles de prédiction de l'âge par la métabolomique constituent des outils prometteurs pour l'étude de la biologie du vieillissement et l'identification des personnes présentant un risque accru de maladies liées à l'âge.
Lien entre les variants pathogènes hétérozygotes et l'évolution de la maladie
Introduction. Bien que les études d'association pangénomique (GWAS) portant sur des variants courants aient permis d'identifier de nombreux loci associés aux taux de métabolites, ces variants ont souvent des effets modestes et ne fournissent pas d'indications directes sur la fonction des gènes. Les variants pathogènes rares identifiés grâce au séquençage de l'exome entier peuvent apporter des informations fonctionnelles plus précises, car ils modifient considérablement l'activité des protéines. Cependant, la plupart des variants rares se présentent sous forme hétérozygote, et leurs conséquences physiologiques ne sont pas encore bien comprises.
Données préliminaires et objectifs de l'étude. Dans cette étude, les chercheurs ont cherché à associer la métabolomique au séquençage de l'exome afin de déterminer de manière systématique comment les variants hétérozygotes rares influencent les taux de métabolites et les traits humains, et de déterminer si ces variants peuvent révéler des effets graduels sur la fonction des gènes et les voies métaboliques [24].
Méthodes. La métabolomique non ciblée et le séquençage de l'exome entier ont été utilisés pour identifier des associations entre les métabolites et les variants pathogènes rares. Les concentrations plasmatiques et urinaires d'un large éventail de métabolites ont été mesurées au sein de vastes cohortes de population, notamment la UK Biobank (UKB) et la German Chronic Kidney Disease (GCKD). Des tests d'agrégation de variants rares ont été réalisés afin d'évaluer si des groupes de variants rares et prédits comme délétères au sein d'un gène étaient associés aux concentrations de métabolites. Des simulations informatiques utilisant des modèles de réseaux métaboliques à l'échelle du génome ont été utilisées pour valider les résultats.
Résultats. 235 associations significatives entre gènes et métabolites, impliquant des variants rares délétères, ont été identifiées dans plusieurs voies métaboliques, dont beaucoup n’avaient jamais été signalées auparavant. Ces associations ont mis en évidence des gènes codant pour des enzymes et des transporteurs qui influencent les concentrations de métabolites dans le plasma ou l’urine. Plusieurs variants génétiques, dont les impacts fonctionnels prédits variaient, ont entraîné des modifications graduelles des taux de métabolites, ce qui a fourni des preuves d’effets dose-dépendants sur la fonction métabolique (figure 45). Il convient de noter que des variants rares dans les gènes des transporteurs de sulfate, tels que SCL13A1 et SLC26A1, étaient fortement associés aux concentrations de sulfate circulant et étaient également liés à la taille et à des traits musculo-squelettiques (Figure 46). L'intégration des données génétiques à la modélisation métabolique computationnelle a confirmé la pertinence fonctionnelle de nombreuses associations.
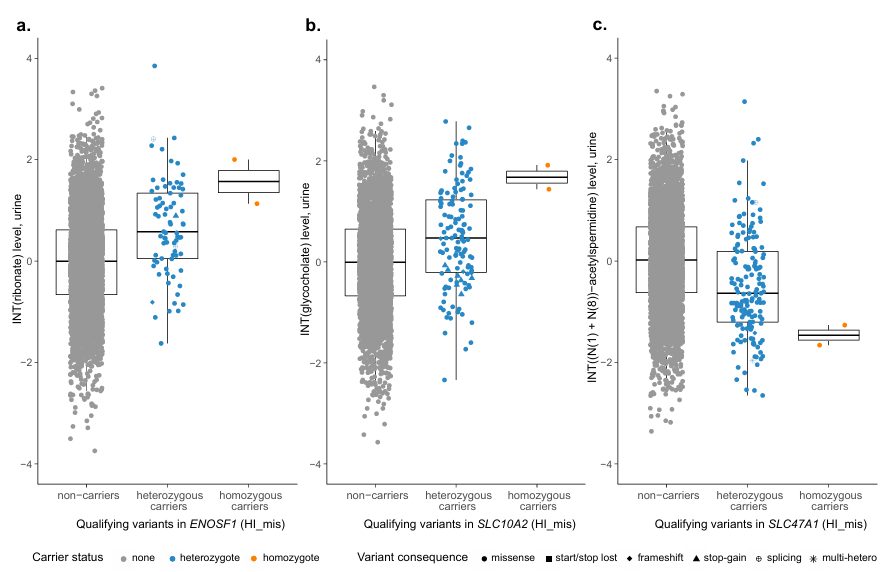
Figure 45. Niveaux de métabolites en fonction du statut de porteur des variants éligibles pour les gènes présentant une association significative et comptant plus d’un porteur homozygote. Les niveaux de métabolites urinaires, après transformation normale inverse et ajustement des covariables, sont représentés sur l’axe des y, tandis que les non-porteurs et les porteurs hétérozygotes des variants éligibles du masque HI_mis sont représentés sur l’axe des x. La couleur et la forme des symboles indiquent respectivement le statut de porteur d’un variant et sa conséquence. Les porteurs de plusieurs variants hétérozygotes éligibles sont signalés par un astérisque. Les boîtes couvrent le 25e au 75e percentile des niveaux de métabolites, la médiane est indiquée par une ligne, et les moustaches s'arrêtent à la dernière valeur observée à une distance de 1,5 fois l'écart interquartile de la boîte. Les niveaux médians de ribonate (n = 4 618) (A), de glycocholate (n = 3 753) (B) et de (N(1) + (N(8))-acétylspermidine (n = 4 619) (C) sont tous plus extrêmes chez les porteurs homozygotes que chez les porteurs hétérozygotes, reflétant un effet dose-réponse. Image reproduite à partir de Scherer et al., Nat Genet, 2025, sous licence CC BY 4.0.
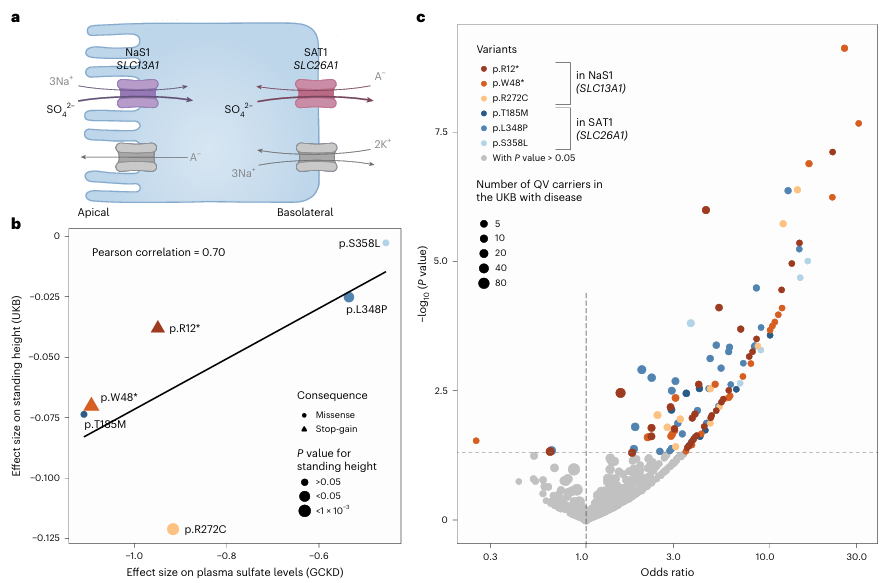
Figure 46. L'impact des variants fonctionnels qualifiants dans les gènes SLC13A1 et SLC26A1 sur la taille, les caractéristiques musculo-squelettiques et les fractures confirme le rôle du sulfate plasmatique en tant qu'indicateur intermédiaire. (A) Représentation schématique du mécanisme de réabsorption du sulfate impliquant NaS1 codé par SLC13A1 au niveau de la membrane apicale et SAT1 codé par SLC26A1 au niveau de la membrane basolatérale des cellules épithéliales. (B) Le nuage de points montre la relation entre l'ampleur de l'effet de six variants qualifiés sur les taux plasmatiques de sulfate dans la cohorte GCKD (axe x) et sur la taille en position debout dans la cohorte UKB (axe des y). (C) Graphique en volcan illustrant les rapports de cotes (axe des x) et les valeurs -log10 (valeurs p) (axe des y) pour les associations des six variants éligibles avec les maladies musculo-squelettiques et les fractures dans l’UKB. Image reproduite à partir de Scherer et al., Nat Genet, 2025, sous licence CC BY 4.0.
Conclusions de l'étude
- La métabolomique a joué un rôle essentiel pour démontrer que même des variants nuisibles hétérozygotes peuvent entraîner des modifications métaboliques mesurables.
- Les effets phénotypiques graduels qui ont été observés montrent comment la variation génétique influence le métabolisme et les caractéristiques humaines.
- Dans l'ensemble, les résultats de cette étude démontrent l'intérêt de la métabolomique pour obtenir des données fonctionnelles permettant d'interpréter les variants génétiques rares, et montrent comment l'association d'un profilage métabolique à grande échelle et du séquençage peut apporter un éclairage nouveau sur le lien complexe entre les variations génétiques et les maladies.
Autres matrices d'échantillonnage
Lait maternel
Introduction. La compréhension du métabolome du lait maternel peut contribuer à éclairer les questions relatives à la nutrition et à la santé des nourrissons ; toutefois, la grande variabilité de la composition du lait d'une personne à l'autre et chez une même personne au fil du temps rend difficile la définition de concentrations de référence pour de nombreux composants. Des études métabolomiques antérieures ont examiné les variations de la composition du lait selon les stades de la lactation, l'état de santé de la mère et les populations, mais relativement peu d'entre elles se sont attachées à identifier les métabolites essentiels systématiquement présents chez des mères issues de milieux divers.
Données préliminaires et objectifs de l'étude. L'objectif de cette étude était de caractériser le métabolome du lait maternel chez des femmes allaitantes en bonne santé issues de milieux divers, afin de définir le lait maternel de manière cohérente chez l'ensemble des mères allaitantes et d'identifier ou de confirmer les éléments essentiels à la nutrition, à la croissance et au développement du nourrisson [25].
Méthodes. Des échantillons de lait maternel ont été prélevés auprès de 31 femmes issues de divers horizons raciaux, ethniques et alimentaires. Les participantes ont également répondu à un questionnaire sur leur état de santé et leur mode de vie. Les échantillons de lait ont été analysés à l'aide d'un profilage métabolomique global, et les relations entre les profils métaboliques et diverses caractéristiques maternelles ou du nourrisson, notamment l'âge de la mère, l'IMC, le nombre d'accouchements, l'âge du nourrisson et les facteurs liés au mode de vie, ont été étudiées. Les voies métaboliques associées aux variables clés ont été identifiées par une analyse d'enrichissement.
Résultats. 389 métabolites étaient présents dans tous les échantillons. En général, l'abondance de ces métabolites communs variait considérablement d'une participante à l'autre. Regroupés par voies métaboliques, les xénobiotiques présentaient la plus grande variabilité au sein de la population étudiée, tandis que les nucléotides étaient les moins variables. L'âge du nourrisson, l'âge de la mère, le nombre de naissances vivantes antérieures et l'IMC avant la grossesse étaient associés à des différences dans le métabolome du lait (figure 47). Les lipides stéroliques et les glucides variaient en fonction de l'âge du nourrisson, le cholestérol et le sulfate de cholestérol augmentant à mesure que les nourrissons grandissaient. L'âge maternel était associé à des changements dans les composés organiques oxygénés, et le nombre de naissances vivantes était lié à des variations dans les acides organiques, les acides nucléiques, les glucides et les lipides.
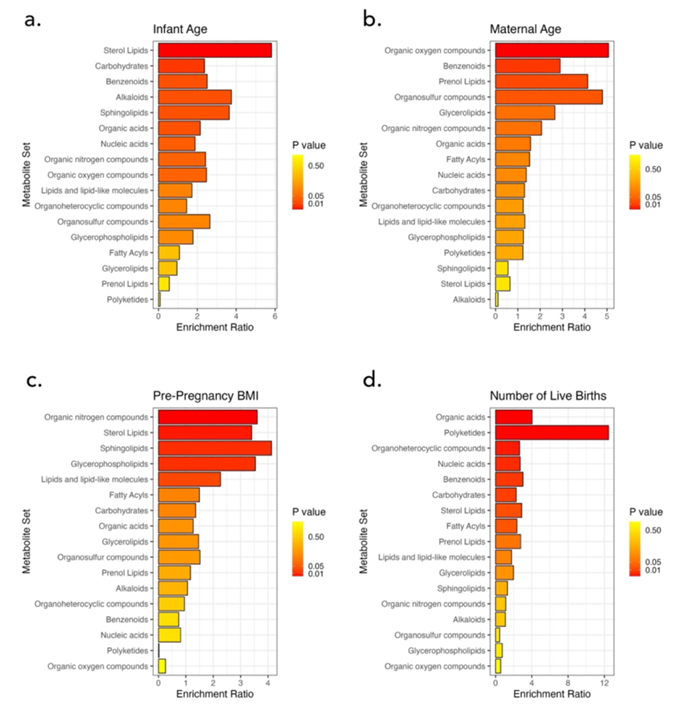
Figure 47. Analyse quantitative de l'enrichissement des ensembles de métabolites (MSEA) en fonction (A) de l'âge du nourrisson, (B) de l'âge de la mère, (C) du nombre de naissances vivantes et (D) de l'IMC avant la grossesse. Image reproduite à partir de Holmes et al., Sci Rep, 2024, sous licence CC BY 4.0.
Conclusions de l'étude
- Cette étude suggère que le lait maternel contient un ensemble fondamental de métabolites conservés qui jouent probablement un rôle essentiel dans la nutrition et le développement psychologique du nourrisson.
- Les métabolites impliqués dans les fonctions cellulaires fondamentales, notamment les nucléotides, le lactose, la créatinine et le glutamate, ont présenté la plus faible variabilité, ce qui suggère que les molécules contribuant à des processus essentiels tels que le métabolisme énergétique font l'objet d'une régulation très stricte.
- Cette étude est un autre exemple qui montre comment la métabolomique peut apporter des éclaircissements scientifiques sur des matrices d'échantillons peu courantes.
Humidité aqueuse
Introduction. L'humeur aqueuse (HA) contient des informations moléculaires précieuses sur les maladies rétiniennes et peut être prélevée de manière non invasive, ce qui en fait une source pratique pour la découverte de biomarqueurs et l'étude des mécanismes pathologiques. Des études antérieures ont eu recours à la métabolomique et à la protéomique pour étudier des maladies oculaires telles que le glaucome, la dégénérescence maculaire néovasculaire liée à l'âge (DMLA néovasculaire) et l'œdème maculaire diabétique (OMD). Cependant, l'interprétation des données moléculaires recueillies à partir de l'H.A. peut être biaisée par des facteurs de confusion, notamment les variations de la concentration totale en protéines et l'état du cristallin (phakique [l'œil contient un cristallin naturel] vs pseudophakique [le cristallin naturel a été remplacé par une lentille artificielle pour traiter la cataracte]).
Données préliminaires et objectifs de l'étude. L'objectif de cette étude était de déterminer l'impact de ces facteurs de confusion oculaires sur les profils protéomiques et métabolomiques de l'AH, à partir d'échantillons provenant de patients atteints de DMLA néovasculaire et d'œdème maculaire diabétique, dans le but à long terme d'améliorer la fiabilité de la découverte de biomarqueurs liés à l'AH [26].
Méthodes. Cette étude clinique prospective et transversale a porté sur 102 participants atteints de DMLA de type exsudatif (nAMD), 18 atteints d’œdème maculaire diabétique (DME) et 18 patients atteints de cataracte sans affection rétinienne, servant de groupe témoin. Des échantillons d’humeur aqueuse (HA) ont été prélevés par paracentèse de la chambre antérieure et congelés en vue de leur analyse. La plateforme d'analyse par extension de proximité Olink Target 96 a été utilisée pour mesurer les protéines liées aux processus inflammatoires, métaboliques, neurologiques et cardiovasculaires. La concentration totale en protéines de l'H.A. a été mesurée séparément. Les métabolites ont été analysés à l'aide Global Discovery Panel Metabolon. Des modèles linéaires avec correction pour les tests multiples ont été utilisés pour identifier les protéines et les métabolites présentant une abondance différentielle entre les groupes, tout en tenant compte des facteurs de confusion oculaires potentiels, notamment l'état du cristallin, la concentration totale en protéines et l'exposition à des médicaments dilatant la pupille. Des analyses de corrélation, le regroupement des profils protéiques et l'enrichissement des ensembles de gènes ont été utilisés pour identifier les voies biologiques associées aux profils moléculaires observés.
Résultats. L'humeur aqueuse (HA) des patients atteints de DMLA néovasculaire (nAMD) et d'œdème maculaire diabétique (DME) présentait des concentrations totales en protéines significativement plus élevées que celles des témoins. Les yeux pseudophaques (lentille artificielle) affichaient des concentrations en protéines de l'HA plus élevées que les yeux phakes (lentille naturelle) et présentaient des taux accrus de protéines liées au remodelage de la matrice extracellulaire et aux voies de signalisation (Figure 48). Une fois les facteurs de confusion pris en compte, le nombre de protéines différentielles entre les groupes de patients et les témoins a considérablement diminué, révélant ainsi des signatures protéiques associées à la maladie et présentant une plus grande pertinence biologique. Des groupes de protéines liés à la signalisation neuronale, aux peptides antimicrobiens, aux enzymes métaboliques et aux réponses au stress oxydatif ont été identifiés et ont montré un enrichissement en protéines précédemment associées à la dégénérescence maculaire liée à l'âge (DMLA) et à l'œdème maculaire diabétique (OMD). Les profils métabolomiques étaient moins affectés par ces facteurs de confusion. L'analyse métabolomique a conduit à la découverte inattendue que les collyres mydriatiques contenant de la phényléphrine/tropicamide étaient associés à une augmentation des taux de plusieurs protéines inflammatoires, en particulier l'interleukine-6 (IL-6), probablement en raison de la présence du métabolite de la phényléphrine, le 3-hydroxymandélate, dans l'AH (Figure 49). La métabolomique a également permis de mieux comprendre les phénotypes de la maladie. Par exemple, des taux élevés de glucose et de fructose et une diminution du 1,5-anhydroglucitol dans les échantillons de DME indiquaient un mauvais contrôle glycémique et une hyperglycémie chronique.
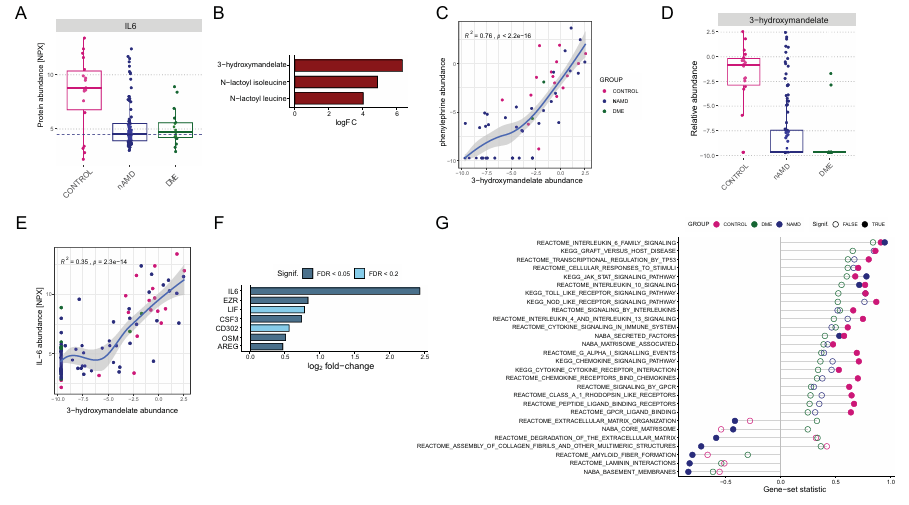
Figure 48. Association entre l'exposition à la phényléphrine/au 3-hydroxymandelate et les profils protéiques de l'AH. (A) Les diagrammes en boîte présentent les valeurs d'abondance relative des protéines (NPX) pour l'IL-6 dans les trois groupes de patients. La ligne pointillée représente le seuil utilisé pour diviser le groupe témoin en vue de l'analyse différentielle d'abondance ultérieure. (B) Diagramme à barres des métabolites significativement associés à des taux élevés par rapport à des taux faibles d'IL-6 au sein du groupe témoin (FDR < 0,05). (C) Corrélation entre les taux de 3-hydroxymandélate et de phényléphrine. (D) Les diagrammes en boîte présentent les valeurs d'abondance relative des métabolites pour le 3-hydroxymandélate dans les trois groupes de patients. (E) Comme en (C), mais pour la corrélation entre les niveaux de 3-hydroxymandélate et d'IL-6. (F) Diagramme à barres des protéines associées à des niveaux élevés par rapport à des niveaux faibles de 3-hydroxymandélate dans le groupe nAMD, avec indication des variations log2 et représentation des niveaux de signification par une échelle de couleurs. (G) Les résultats de l'analyse GSEA pour les taux élevés par rapport aux taux faibles de 3-hydroxymandélate dans le groupe nAMD sont présentés. Image reproduite à partir de Titz et al., Transl Vis Sci Technol, 2024, sous licence CC BY 4.0.

Figure 49. Influence du statut cristallinien sur les profils protéiques de l'humeur aqueuse. (A) Carte thermique illustrant l'abondance différentielle des protéines, ajustée en fonction de la concentration protéique, entre les yeux pseudophaques et phakes au sein des groupes de patients atteints de DMLA néovasculaire (nAMD) et d'œdème maculaire diabétique (DME). Les variations log2 sont représentées par un dégradé de couleurs, et les protéines présentant des différences statistiquement significatives sont signalées (*P < 0,05, ajusté selon le FDR). La carte thermique se limite aux 15 protéines les mieux classées selon la valeur absolue du logFC. (B) Présentation des résultats de l'analyse GSEA pour l'état du cristallin (pseudophaque vs phakique). Image reproduite à partir de Titz et al., Transl Vis Sci Technol, 2024, sous licence CC BY 4.0.
Conclusions de l'étude
- Les résultats de cette étude montrent que l'analyse protéomique de l'AH peut être fortement influencée par des facteurs de confusion oculaires et liés à la procédure, mais que la métabolomique y est moins sensible.
- Dans cette étude, la métabolomique a permis d'identifier les collyres mydriatiques contenant de la phényléphrine comme un facteur de confusion jusque-là méconnu. Le 3-hydroxymandélate présent dans ces collyres a été associé à une augmentation des taux de protéines inflammatoires. Non seulement cela a permis de mettre au jour un nouveau mécanisme dans la biologie oculaire, mais cela montre également que les études futures devront tenir compte de l'exposition à la phényléphrine afin d'interpréter avec précision les données omiques de l'hypertension artérielle.
- Cette étude illustre également comment la métabolomique peut fournir des informations scientifiques cruciales à partir d'une matrice d'échantillons inhabituelle.
Prélèvements cervicaux
Introduction. Le microbiote vaginal joue un rôle essentiel dans la santé reproductive des femmes. Un milieu vaginal sain est généralement dominé par des espèces de Lactobacillus, en particulier Lactobacillus crispatus, qui sont associées à des effets protecteurs, tandis que des communautés microbiennes plus diversifiées et riches en anaérobies sont liées à la vaginose bactérienne (VB). Lactobacillus iners joue un rôle paradoxal dans la VB car, contrairement aux autres espèces de Lactobacillus, il est associé à de moins bons résultats cliniques et à un risque accru de transition vers la VB, bien qu'il soit l'organisme dominant chez la plupart des femmes. Le traitement standard de la VB par le métronidazole aboutit souvent à des communautés dominées par L. iners, qui sont instables et sujettes à des rechutes. Pour relever ce défi clinique, il faut de nouvelles stratégies capables de moduler le microbiome vaginal afin de favoriser la croissance d’un microbiote durable et bénéfique pour la santé.
Données préliminaires et objectifs de l'étude. La biologie de L. iners est mal comprise, principalement parce qu'il est difficile de le cultiver et de l'étudier in vitro. Comparé à d'autres espèces de Lactobacillus, il possède un génome plus petit et une capacité métabolique limitée, ce qui suggère une dépendance vis-à-vis des nutriments externes. Sur la base de ces résultats antérieurs, l'objectif de cette étude était d'identifier les principales dépendances métaboliques de L. iners et de déterminer si cette vulnérabilité pouvait être ciblée pour faire évoluer le microbiote vaginal vers des compositions plus bénéfiques et améliorer les résultats du traitement de la VB [27].
Méthodes. Des isolats cliniques ont été prélevés auprès d'une vaste cohorte de femmes sud-africaines et américaines comprenant à la fois des cas de VB et des cas sans VB. Un catalogue génomique exhaustif a été constitué à partir de ces isolats afin de permettre la comparaison des capacités métaboliques entre les différentes espèces microbiennes. Les échantillons de lavage cervico-vaginal ont été analysés par métabolomique globale et la composition du microbiote a été caractérisée par séquençage du gène de l'ARNr 16S. Des analyses statistiques ont été utilisées pour corréler les niveaux de métabolites avec la structure de la communauté bactérienne (par exemple, communautés dominées par Lactobacillus vs communautés associées à la VB).
Résultats. Les résultats ont révélé que la croissance de L. iners dépendait exclusivement de la cystéine. Une croissance vigoureuse de diverses souches de L. iners a été observée dans des milieux de culture enrichis en L-cystine, tandis qu'aucune croissance n'a été constatée dans les cultures pauvres en L-cystine. Il a été démontré que L. iners avait une capacité limitée à utiliser des sources complexes de cystéine, ce qui explique ses besoins spécifiques en matière de croissance et met en évidence une vulnérabilité métabolique majeure. Dans les échantillons cliniques, les taux de cystéine étaient significativement plus élevés chez les femmes ne présentant pas de vaginose bactérienne (VB) et étaient en corrélation positive avec l'abondance de Lactobacillus, tandis qu'ils étaient en corrélation négative avec les bactéries associées à la VB (Figure 50). Des analyses génomiques ont révélé que L. iners ne possède pas bon nombre des systèmes de transport de la cystéine et des molécules contenant de la cystéine présents chez d'autres espèces de Lactobacillus, ce qui suggère qu'il a une flexibilité métabolique limitée. Les inhibiteurs de l'absorption de la cystine ont sélectivement supprimé la croissance de L. iners sans affecter de manière significative les autres espèces de Lactobacillus, et en compétition
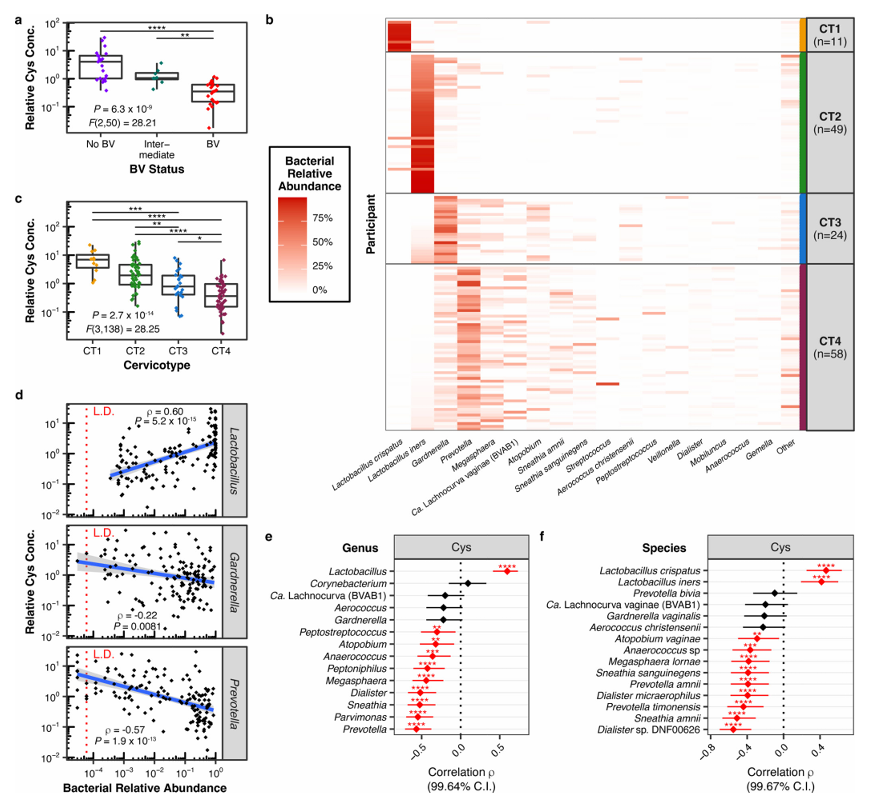
Figure 50. Les concentrations vaginales de cystine (Cys) sont plus élevées chez les femmes ne présentant pas de vaginose bactérienne (VB) et sont corrélées à un microbiote dominé par les Lactobacillus. (A) Concentration relative de Cys en fonction du statut VB dans le liquide de lavage cervico-vaginal (CVL) de 53 femmes sud-africaines (21 sans VB, 24 avec VB et 8 intermédiaires). (B) Composition du microbiote bactérien FGT chez 142 femmes sud-africaines non infectées par le VIH (dont les 53 de la figure A), déterminée par séquençage du gène 16S rRNA bactérien. (C) Concentrations relatives de Cys par CT dans le liquide cervical (CTL) des femmes de la figure (B). (D) Corrélation de Spearman bilatérale entre les concentrations de Cys et les abondances bactériennes relatives des genres Lactobacillus, Gardnerella et Prevotella. (E, F) Coefficients de corrélation de Spearman bilatéraux (ρ) avec intervalles de confiance ajustés entre les concentrations de Cys et les abondances relatives de chaque genre (E) ou espèce (F) détectés à une prévalence > 50 % dans la cohorte (n = 142). Image reproduite à partir de Bloom et al., Nat Microbiol, 2022, sous licence CC BY 4.0.
Conclusions de l'étude
- Cette étude a mis en évidence la dépendance à la cystéine comme une caractéristique biologique déterminante de L. iners, ce qui permet d'expliquer les difficultés liées à la culture de cette espèce.
- L. iners présente une capacité particulièrement limitée à transporter et à utiliser diverses sources de cystéine, ce qui le rend particulièrement dépendant de certaines formes spécifiques de cystéine exogène. Cette contrainte métabolique contribue à expliquer son comportement écologique et le distingue d'espèces davantage associées à la santé, telles que L. crispatus.
- Les résultats de l'étude suggèrent également que les taux de cystéine provenant de l'hôte pourraient déterminer si ce sont les communautés dominées par les Lactobacillus ou celles associées à la vaginose bactérienne qui prédominent, ce qui laisse penser que la disponibilité des métabolites pourrait être un facteur déterminant de la structure de la communauté microbienne dans l'environnement vaginal.
- Dans l'ensemble, cette étude a démontré la validité du concept selon lequel les inhibiteurs de l'absorption de la cystine pourraient supprimer activement L. iners et renforcer l'efficacité du métronidazole, ce qui laisse entrevoir une nouvelle approche pour le traitement de la vaginose bactérienne.
Liquide synovial
Introduction. L'arthrose touche des millions de personnes et contribue de manière significative à l'invalidité. L'arthrose post-traumatique (APT) de la cheville revêt une importance particulière, car elle touche souvent des personnes plus jeunes et peut se développer des années après la blessure initiale. Malgré sa prévalence, les processus biologiques précoces à l'origine de l'APT restent mal compris, et les méthodes diagnostiques standard, telles que l'imagerie et les examens cliniques, ne permettent pas de détecter les modifications précoces de la maladie, ce qui limite les possibilités d'intervention précoce. Une lésion articulaire aiguë déclenche des réponses inflammatoires dans le liquide synovial, notamment une élévation des cytokines, des métalloprotéinases matricielles et des lésions cellulaires, qui peuvent toutes contribuer à la dégradation du cartilage et à la dégénérescence articulaire à long terme. Le liquide synovial sert de réservoir aux métabolites dérivés des tissus articulaires, et des études antérieures suggèrent que la composition lipidique change en cas de lésion, d’inflammation et d’arthrite. Cependant, comme les travaux antérieurs s’appuyaient sur des techniques analytiques relativement limitées, l’ensemble des altérations métaboliques qui se produisent après une lésion reste mal compris.
Données préliminaires et objectifs de l'étude. L'objectif de cette étude était d'utiliser la métabolomique globale pour identifier les modifications métaboliques liées aux lipides associées à une lésion aiguë et aux processus pathologiques précoces, dans le but à long terme de découvrir des biomarqueurs et des cibles thérapeutiques potentielles susceptibles d'améliorer la compréhension et la prise en charge de l'arthrose précoce du genou [28].
Méthodes. L'étude consistait en une analyse de cohorte rétrospective portant sur des patients présentant des fractures intra-articulaires unilatérales de la cheville nécessitant une intervention chirurgicale. Vingt patients ont été inclus, et du liquide synovial (LS) a été prélevé à la fois sur la cheville blessée et sur la cheville controlatérale non blessée au moment de l'intervention chirurgicale. Un sous-groupe de sept patients a également subi un deuxième prélèvement bilatéral de LS six mois plus tard. Les concentrations de métabolites ont été corrigées pour tenir compte de la dilution survenue lors du prélèvement par lavage, à l'aide d'une méthode de normalisation basée sur l'urée qui comparait les taux d'urée dans le liquide synovial à ceux du sérum. Après établissement d'un profil métabolomique global, une classification par forêt aléatoire a été utilisée pour identifier les métabolites clés permettant de différencier le LS de la cheville blessée du LS témoin. Des analyses de corrélation ont été réalisées afin d'identifier les liens biologiques entre les métabolites lipidiques, les cytokines inflammatoires et les métalloprotéases matricielles.
Résultats. Les acides gras à longue chaîne, les acides gras polyinsaturés (AGPI), les sphingomyélines et les lysolipides présentaient des taux significativement plus élevés dans les chevilles fracturées par rapport aux articulations controlatérales saines. Une analyse par forêt aléatoire a confirmé que les acides gras et les lysolipides constituaient les principaux facteurs permettant de distinguer les articulations lésées des articulations saines (figure 51). Au fil du temps, bon nombre de ces anomalies lipidiques ont partiellement disparu, plusieurs acides gras et sphingomyélines ayant considérablement diminué six mois après l'intervention chirurgicale. De même, la capacité des profils métabolomiques à distinguer les articulations blessées des articulations témoins a diminué au bout de six mois, ce qui suggère que la réponse métabolique aiguë à la blessure s'estompe avec le temps. Les métabolites lipidiques ont montré de fortes corrélations positives avec les cytokines inflammatoires et les métalloprotéases matricielles, établissant un lien entre la dérégulation lipidique, l'inflammation et les processus de dégradation tissulaire (Figure 52). L'ampleur et l'étendue des modifications lipidiques étaient associées à la gravité de la lésion, les fractures plus graves présentant des élévations plus importantes des acides gras, des lysolipides et des métabolites associés.
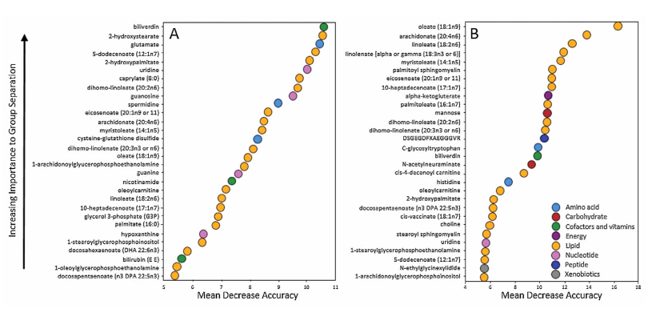
Figure 51. Analyse de classification par la méthode Random Forest comparant le groupe « blessé » au groupe « témoin controlatéral » au début de l'étude (A), et le groupe « blessé » au début de l'étude au groupe « blessé » à 6 mois (B). Image reproduite à partir de Leimer et al., J Orthop Res, 2017, sous licence CC BY 4.0.
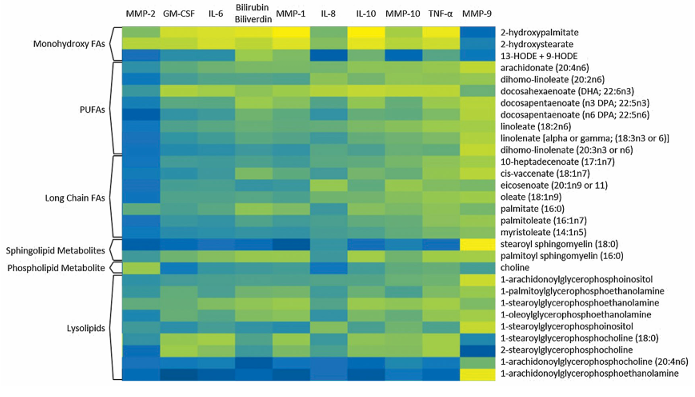
Figure 52. Corrélations entre les métabolites lipidiques et les taux de cytokines chez une même cohorte de patients. Les zones de la carte thermique vont du noir au bleu puis au jaune, le noir représentant les corrélations les plus faibles et le jaune les plus fortes. Toutes les valeurs de corrélation significatives étaient positives. Image reproduite à partir de Leimer et al., J Orthop Res, 2017, sous licence CC BY 4.0.
Conclusions de l'étude
- Les résultats de cette étude suggèrent qu'une fracture intra-articulaire aiguë de la cheville entraîne l'apparition d'une signature lipidique distincte, liée à la lésion, dans le liquide synovial, caractérisée par une augmentation des acides gras libres, des lysolipides et des sphingolipides.
- Les fortes corrélations entre les métabolites lipidiques et les cytokines ou les métalloprotéases matricielles suggèrent que ces altérations lipidiques sont étroitement liées à l'inflammation, aux lésions tissulaires et à la dégradation du cartilage dans l'articulation lésée.
- Étant donné que de nombreuses modifications lipidiques induites par la lésion étaient transitoires et revenaient à la normale dans les six mois suivant l'intervention chirurgicale, la réponse métabolique pourrait s'inscrire dans le cadre d'un processus aigu de lésion et de guérison ; par ailleurs, les élévations précoces de ces métabolites pourraient également déclencher des voies de signalisation contribuant à la dégénérescence articulaire à long terme et à l'évolution vers l'arthrose post-traumatique.
- Les métabolites lipidiques pourraient servir de biomarqueurs d'une lésion articulaire précoce et du risque d'arthrose post-traumatique, ainsi que de cibles potentielles pour une intervention.
Points à retenir de ce chapitre
- La métabolomique peut être appliquée à de nombreux types d'études et à un large éventail de thèmes dans le domaine de la recherche fondamentale.
- Dans la plupart des cas, la métabolomique est essentielle pour acquérir une compréhension fondamentale ou faire une découverte cruciale qui fait progresser la recherche et lui confère une importance accrue.

Télécharger le guide complet au format PDF
Téléchargez ce guide complet conçu pour vous apprendre les tenants et les aboutissants de l'un des outils omiques les plus puissants de la boîte à outils de tout scientifique : la métabolomique.
Télécharger maintenantNous contacter
Parler avec un expert
Demandez un devis pour nos services, obtenez plus d'informations sur les types d'échantillons et les procédures de manipulation, demandez une lettre de soutien ou posez une question sur la façon dont la métabolomique peut faire avancer votre recherche.
Siège social
617 Davis Drive, Suite 100
Morrisville, NC 27560
Adresse postale :
P.O. Box 110407
Research Triangle Park, NC 27709
+1 (919) 572-1711
Siège international
Metabolon GmbH
Zeppelinstraße 3
85399 Hallbergmoos
Allemagne